

Key Points
Fetal and adult lung megakaryocytes are transcriptionally and phenotypically distinct from fetal liver and bone marrow megakaryocytes.
Lung megakaryocytes have an immune signature and may be primed for more efficient platelet production.

Abstract
Megakaryocytes (MKs) are responsible for platelet biogenesis, which is believed to occur canonically in adult bone marrow (BM) and in the fetal liver during development. However, emerging evidence highlights the lung as a previously underappreciated residence for MKs that may contribute significantly to circulating platelet mass. Although a diversity of cells specific to the BM is known to promote the maturation and trafficking of MKs, little investigation into the impact of the lung niche on the development and function of MKs has been done. Here, we describe the application of single-cell RNA sequencing, coupled with histological, ploidy, and flow cytometric analyses, to profile primary MKs derived from syngeneic mouse lung and hematopoietic tissues. Transcriptional profiling demonstrated that lung MKs have a unique signature distinct from their hematopoietic counterparts, with lung MKs displaying enrichment for maturation markers, potentially indicating a propensity for more efficient platelet production. Reciprocally, fetal lung MKs also showed the robust expression of cytokines and growth factors that are known to promote lung development. Lastly, lung MKs possess an enrichment profile skewed toward roles in immunity and inflammation. These findings highlight the existence of a lung-specific MK phenotype and support the notion that the lung plays an independent role in the development and functional maturation of MKs. The immune phenotype displayed by lung MKs also introduces their potential role in microbial surveillance and antigen presentation.
Introduction
Megakaryocytes (MKs) and the platelets that they produce are essential for clot formation, but both are also involved in critical biological processes, including development, inflammation, and regeneration.1-4 The majority of platelet biogenesis is believed to occur canonically in the adult bone marrow (BM) and in the fetal liver during development.4 As such, our current understanding of MK development stems predominantly from the investigation of these tissues. The presence of MKs in the lung was first described in 1893, and emerging evidence further highlights the lung as a previously underappreciated residence for MKs that may contribute significantly to circulating platelet mass.5-14 Interest in lung MKs was reinvigorated with the application of intravital 2-photon microscopy to the lungs of live mice, showing de novo platelet production and immobile MKs residing in the lung interstitium.15 However, approximations of the absolute number of platelets being generated in the lung remain heavily debated. Residency in the lung suggests that MKs are interacting with a diversity of cells that differ widely from those found in the BM. Various cells specific to the BM niche, including osteoblasts and mesenchymal stromal cells, are known to promote the maturation and trafficking of MKs to carry out platelet production.16-24 Beyond this, the BM niche induces MKs to adopt a myriad of other functions, including BM homeostasis and viral immunity.1,25,26
Therefore, we hypothesized that MK maturation in the context of the lung niche induces localized MKs to adopt a lung-specific phenotype and functional profile. The need to better understand megakaryopoiesis in this context is further highlighted by recent work demonstrating the diverse lung-specific influence of platelets. The multifaceted role of platelets in the lung in health and disease includes support of embryonic pulmonary development, protection against bacterial exotoxin-induced damage to the lung epithelium, and promotion of pulmonary fibrosis.27-31 Whether these platelets are sourced from MKs found and functionalized in the lung or BM is not known.
The role of platelets in inflammation and immunity is well described and continues to be a critical point of interest because of the implications for human health.3,32-34 Although MKs alone are known to express functional Toll-like receptors (TLRs) and produce inflammatory cytokines, our knowledge of the MK immune profile largely stems from in vitro studies, limiting our understanding of the significance of in vivo MKs in immunity.1 Furthermore, involvement of BM MKs in immunity is primarily only appreciable upon stimulation by a stressor, such as infection or sepsis.26,35-37 In contrast, recent evidence suggests that, under homeostatic conditions, lung MKs may possess a transcriptional signature skewed toward innate immune functions.15 Lung MKs also have demonstrated responsiveness to pulmonary insults, including bacterial pneumonia and bleomycin.15,38
Importantly, the methodologies used in previous studies have significant limitations with regard to the tracking and isolation of lung MKs. These include the use of promiscuous markers of the MK lineage,39-41 a lineage tracing model with significant off-target recombination in immune cell populations,42,43 as well as the use of bulk sequencing methodologies that do not allow for the identification and investigation of individual cells.
MKs are rare cells, making up just 0.1% of the mouse BM, and there is a paucity of useful reagents/markers to isolate and study MK development. This led us to choose single-cell RNA Sequencing (scRNA-Seq) to transcriptionally profile lung MKs at single-cell resolution.44,45 Furthermore, to profile MKs at different stages of development, we assessed fetal and adult MKs. Flow cytometry, in combination with scRNA-Seq analyses, demonstrated that the majority of lung MKs are terminally mature and possess a transcriptional and phenotypic profile that suggests they are primed for efficient platelet production. This analysis also confirmed the notion that lung MKs have a phenotype skewed toward immunity and inflammation. These findings demonstrate the existence of a unique lung MK phenotype and support the hypothesis that the lung microenvironment plays a critical role in the development and functionalization of MKs. The immune phenotype displayed by lung MKs also introduces their potential role as an integral component in the immune landscape of the lung.
Materials and methods
Mice
Ten-week-old and timed-pregnant C57BL/6 mice were purchased from The Jackson Laboratory. All animal housing and experimental procedures were approved by the Boston University School of Medicine Institutional Animal Care and Use Committee.
Tissue isolation and processing
Fetal tissue processing.
Twenty embryonic day 13 (E13) fetal mice were isolated from 3 timed-pregnant mice. The fetal liver and lung were isolated from surrounding tissue by blunt dissection and set aside in a solution of 10% characterized fetal bovine serum in Hanks balanced salt solution (HBSS; Gibco). Using a 5-mL syringe fitted with a 16-gauge needle, the fetal liver was drawn up and expelled several times to dissociate the tissue. Digested livers were filtered through a 70-µm cell strainer before being resuspended in 3 mL of Red Blood Cell (RBC) Lysis Buffer (Sigma) for 5 minutes at 37°C. For all samples, 70-µm strainers were used for filtering to best accommodate the large size of MKs, which average ∼30 µm in diameter in mice and can be as large as 60 µm.46,47 Post-RBC lysis, cells were washed, filtered, and resuspended in sort buffer (phosphate-buffered saline [PBS] with 2% RNase, DNase-free; Millipore). Fetal lungs were minced gently with a scalpel and placed into a digestion buffer consisting of HBSS, 2.4 U/mL Dispase II (Roche), 0.1% Collagenase A (Roche), and 2.5 mM CaCl2 (Fluka). Four milliliters of digestion buffer was used per 5 embryonic lungs. This digestion mixture was placed on a rocker at 37°C for 1 hour. Postdigestion, lung samples were filtered and resuspended in RBC Lysis Buffer for 1 minute at 37°C and then washed, filtered, and resuspended in sort buffer.
Adult tissue processing.
Week-10 adult C57BL/6 mice (3 females and 2 males) were anesthetized with isoflurane and euthanized by cervical dislocation. Using scissors, the abdomen was opened, and the renal and abdominal artery were cut. A thoracotomy was performed to expose the heart and lungs, and mice were perfused via the right ventricle with 5 mL of PBS. Postperfusion, lungs were removed and minced with a scalpel before being placed in a digestion buffer containing the following: HBSS, 1 mg/mL DNase I (Sigma), 0.5 mg/mL Liberase (Sigma), and 1 mg/mL Elastase (Worthington). Six milliliters of digestion buffer were used per adult lung. This digestion mixture was placed on a rocker at 37°C for 30 minutes. Postdigestion, lung samples were pipetted up and down with a 5-mL serological pipette to dissociate tissue; samples were filtered and resuspended in 3 mL of RBC Lysis Buffer for 5 minutes at 37°C. BM was isolated from the femur and tibia as previously described.48 In short, skin and muscle were dissected away from the femur and tibias bilaterally. Bones were crushed by mortar and pestle to expose the marrow and then flushed with PBS. BM isolates were filtered and resuspended in RBC Lysis Buffer and washed, filtered, and resuspended in sort buffer.
Tissue histology
Adult mice were euthanized and transcardially perfused as described above. Postperfusion, lungs were removed and inflated via the trachea with 4% paraformaldehyde to fix inflate lungs. Fetal lungs could not be perfused and, thus, were placed directly into fixative. Lungs were fixed and cryoprotected overnight in sucrose and paraformaldehyde. Fixed tissue was embedded and frozen in Optimum Cutting Temperature embedding medium. Five- to 6-µm tissue sections were made using a cryostat and placed on slides. Sections were rinsed with PBS prior to permeabilizing and blocking in a solution of 0.4% Triton X-100 and 10% normal donkey serum. Sections were stained overnight using the following primary antibodies: rat anti-mouse CD41 (1:100, MWReg30; BioLegend), Armenian hamster anti-mouse CD42d (1:100, 1C2; BioLegend), rabbit anti-TTF1/NKX2.1 (1:200, EP1584Y; Abcam), Syrian hamster anti-PDPN (1:100, 8.1.1; eBioscience), and goat anti-mouse VECAD (1:100). Sections were subsequently stained with the following secondary antibodies (Jackson ImmunoResearch, all at 1:400 dilution): donkey anti-rat Alexa Fluor 488, donkey anti-rabbit Alexa Fluor 647, goat anti–Syrian hamster Alexa Fluor 647, donkey anti–Armenian hamster Cy3, and donkey anti-goat Cy3. Sections were counterstained with Hoechst stain, coverslipped, and imaged on a Zeiss LSM 710-Live Duo Confocal microscope. Postimage processing was performed using ImageJ (National Institutes of Health).
Flow cytometry
A solution of 1% bovine serum albumin (BSA) in PBS, 5 mM EDTA was used for staining and washing cells. Isolated cells were resuspended with the appropriate antibodies and stained on ice for 30 minutes. Fc-receptor block treatment was performed using an anti-mouse CD16/32 antibody (clone 93; BioLegend) for 10 minutes prior to fluorescent staining. Flow cytometry was conducted on a Stratedigm S1000EXI, and FlowJo v10.6.2 software was used for analysis. The following antibodies were used: CD41a-BV421 (MWReg30; BioLegend), CD41a-PE (MWReg30; BioLegend), CD41a-APC (MWReg30; BioLegend), CD42d-APC (1C2; eBioscience), CD42d-PE (1C2; eBioscience), CD42d–Alexa Fluor 488 (1C2; eBioscience), CD284/TLR4-APC (SA15-21; BioLegend), CD282/TLR2-APC (QA16A01; BioLegend), I-A/I-E/MHCII-APC (M5/114.15.2; BioLegend), and CD74–Alexa Fluor 647 (In1; BioLegend).
Ploidy assay.
Pelleted cells from a BSA gradient were isolated and stained with CD41a-APC for 30 minutes on ice. Stained cells were subsequently washed and resuspended in a solution of 0.5 mg/mL propidium iodide, 10% NP40, and 1% BSA in PBS and then immediately analyzed by flow cytometry.
BSA enrichment.
scRNA-Seq
Cell sorting for scRNA-Seq.
Cell sorting was conducted on a Beckman Coulter MoFlo Astrios. Isolated primary cells were stained with CD41a-APC and resuspended in sort buffer (2% BSA in PBS) containing Calcein Blue AM (1:1000). Live singlets and the top 1% of CD41a-APC+ events were sorted into sort buffer. Cells counts and viability after sorting were confirmed using a hemocytometer and trypan blue.
10x Chromium.
Isolated single cells were captured using the 10x Genomics Chromium platform and prepared using a Single Cell 3′ v3 kit. Library preparation and sequencing were done at the Boston University Microarray and Sequencing Resource Core using an Illumina NextSeq 500 System.
Bioinformatics analysis
Reads were demultiplexed and aligned to the mouse genome assembly (GRCm38; Ensembl) with the Cell Ranger pipeline v.3.0.2 (10x Genomics). Further analyses were done using Seurat version 3.1.4.51 After inspection of the quality control metrics, cells with >12% of mitochondrial content or <800 detected genes were excluded for downstream analyses. We normalized and scaled the unique molecular identifier (UMI) counts using the regularized negative binomial regression (sctransform).52 Following the standard procedure in Seurat’s pipeline, we performed linear dimensionality reduction (principal component analysis) and used the top 20 principal components to compute the unsupervised Uniform Manifold Approximation and Projection (UMAP)53 and the clusters (Louvain method),54 which were computed at a range of resolutions from 1.5 to 0.05 (more to fewer clusters). Adult populations were annotated using Louvain resolution 0.75, and fetal populations were annotated using Louvain resolution 0.25. The MK populations in adult BM and lung and fetal liver and lung were combined for subsequent analyses (Figure 2). Cell cycle scores and classifications were done using the Seurat method.55 To annotate our samples, the same scoring method was used to calculate the enrichment in molecular signatures derived from cell types present in a previous publication.56 The cutoffs for independent filtering57 prior to differential expression testing required that genes were detected in ≥10% of the cells of either population and had a natural log fold-change ≥0.25 between populations. The tests were performed using Seurat’s wrapper for the MAST framework.58 For a comparison of the performance of methods for single-cell differential expression, see the article by Soneson and Robinson.59
Statistical analysis
Significance for Enrichr-based pathway enrichment was determined by Fisher’s exact test.60,61 Significance for pairwise scRNA-Seq gene expression comparisons was determined using the MAST framework.58 Significance for flow cytometry assays performed in triplicate was determined by the Student paired t test.
Results
Adult lung MKs are localized to the alveolar interstitium of the distal lung, and fetal lung MKs are found intra- and extravascularly
To better understand the location and the potential cellular interactions that may promote the development of a lung MK phenotype, immunofluorescent microscopy was performed on cryosections of fetal and adult mouse lungs to visualize the niche of lung MKs. Adult lung MKs are preferentially localized to the distal lung within the interstitial space between adjacent alveoli (Figure 1A-C). Additionally, multinucleated MKs were often found in close proximity to distal lung epithelial cells expressing NKX2.1 and podoplanin. Fetal lung MKs are found in the developing lung intra- and extravascularly (Figure 1D-E). Platelets were notably less abundant in fetal lung sections.
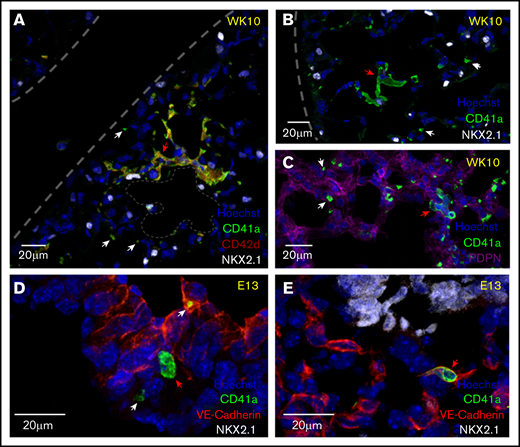
Adult lung MKs are localized to the alveolar interstitium of the distal lung, and fetal lung MKs are found intra- and extravascularly. (A-C) Immunofluorescent staining of 5- to 6-µm fixed-frozen week-10 (WK10) adult mouse lung sections stained for markers of MKs (CD41a and CD42d), lung epithelial cells (NKX2.1), alveolar type 1 epithelial cells (PDPN), and nuclei (Hoechst). Lung borders and an alveolar airspace are outlined with gray dashes. Multinucleated adult lung MKs were preferentially localized to the interstitium of the alveolar septum between neighboring alveoli. (D-E) Immunofluorescent staining of 5- to 6-µm fixed-frozen E13 fetal mouse lung sections stained for VE-Cadherin (endothelial cell marker), CD41a, NKX2.1, and Hoechst. Fetal lung MKs were identified in the developing lung in extravascular (D) and intravascular (E) spaces. Platelets were notably less prevalent in the fetal lung. White and red arrows highlight platelets and MKs, respectively.
Adult lung MKs are localized to the alveolar interstitium of the distal lung, and fetal lung MKs are found intra- and extravascularly. (A-C) Immunofluorescent staining of 5- to 6-µm fixed-frozen week-10 (WK10) adult mouse lung sections stained for markers of MKs (CD41a and CD42d), lung epithelial cells (NKX2.1), alveolar type 1 epithelial cells (PDPN), and nuclei (Hoechst). Lung borders and an alveolar airspace are outlined with gray dashes. Multinucleated adult lung MKs were preferentially localized to the interstitium of the alveolar septum between neighboring alveoli. (D-E) Immunofluorescent staining of 5- to 6-µm fixed-frozen E13 fetal mouse lung sections stained for VE-Cadherin (endothelial cell marker), CD41a, NKX2.1, and Hoechst. Fetal lung MKs were identified in the developing lung in extravascular (D) and intravascular (E) spaces. Platelets were notably less prevalent in the fetal lung. White and red arrows highlight platelets and MKs, respectively.
Creation of a single-cell transcriptomic atlas of primary fetal and adult murine MKs from hematopoietic and pulmonary tissues
The developmental trajectory of the hematopoietic and pulmonary systems involves many stages throughout embryonic and postnatal maturation.45,62,63 To create a transcriptomic library that spans multiple stages and sites of MK development, scRNA-Seq was performed on cells isolated from fetal liver and lung, as well as from adult BM and lung (supplemental Figure 1A). E13 was chosen as the fetal time point, which is when the majority of murine hematopoietic development is occurring in the liver.49,62 Notably, postnatal lung development continues for many weeks but begins to plateau at around week 8 in adult mice.64 To ensure sampling of adult MKs from a developmentally stable source, week 10 was chosen as the adult time point. Enrichment of MKs in all tissues was accomplished using flow cytometry to sort for CD41a+ cells (supplemental Figure 1B-C). No BSA enrichment was done prior to sorting. Because CD41a is broadly expressed throughout the MK lineage, it was chosen to ensure that MKs at various stages of development were being captured. Furthermore, based on prior studies that have assessed CD41a expression across hematopoietic cell types, we chose to gate on the top 1% of CD41a+ events to enhance enrichment of MK-lineage cells while still allowing for the capture of MK progenitors.39,40,65 The gating strategy used, combined with the single-cell transcriptomic profiling, provides sufficient resolution to ensure accurate, yet broad, analysis of MKs at all development stages. Sorted MKs were also imaged to ensure that the strategy used successfully captured nucleated MKs across a range of developmental states and not platelet aggregates (supplemental Figure 2).
Lung MKs display a distinct transcriptional profile that is enriched for markers of maturation
UMAP clustering was performed on cells isolated from fetal lungs and livers, as well as from adult lungs and BM (supplemental Figure 3). Fetal and adult MK-lineage clusters were selected based on the expression of key MK-lineage marker genes, including Fli1, Pf4, and Itga2b (Figure 2B,D, dashed box), and remapped (Figure 2A,C). Fetal lung MKs clustered separately from fetal liver MKs (Figure 2A). This trend was also seen when comparing adult lung MKs (ALu) with adult BM MKs (ABM-1, ABM-2, and ABM-3) (Figure 2C), highlighting the unique transcriptional profile of fetal and adult lung MKs. It should be noted that intravascular staining was not used prior to scRNA-Seq. However, other investigators have demonstrated that ∼80% of total lung MKs are extravascular, and our UMAP clustering resulted in 1 fetal lung cluster and 1 adult lung cluster.15 This suggests that the transcriptional profile of lung MKs is relatively homogenous compared with BM and fetal liver MKs.

Lung MKs are transcriptionally distinct from their hematopoietic counterparts. (A,C) UMAP clustering of MK-lineage clusters from fetal liver (FLiv-1, FLiv-2), fetal lung (Flu), adult BM (ABM-1, ABM-2, ABM-3), and ALu. Fetal and adult lung MKs clustered independently from their hematopoietic counterparts. Numbers in parentheses indicate cell counts for associated clusters. (B,D) Violin plots comparing expression of key MK genes in associated clusters. Key genes used to confirm MK lineage identity are outlined with a dashed box. Colors of violin plots in panels B and D correlate with their respective clusters shown in panels A and C, respectively. *P < .01. (Asterisks indicating significance were withheld from panel B for visual clarity.)
Lung MKs are transcriptionally distinct from their hematopoietic counterparts. (A,C) UMAP clustering of MK-lineage clusters from fetal liver (FLiv-1, FLiv-2), fetal lung (Flu), adult BM (ABM-1, ABM-2, ABM-3), and ALu. Fetal and adult lung MKs clustered independently from their hematopoietic counterparts. Numbers in parentheses indicate cell counts for associated clusters. (B,D) Violin plots comparing expression of key MK genes in associated clusters. Key genes used to confirm MK lineage identity are outlined with a dashed box. Colors of violin plots in panels B and D correlate with their respective clusters shown in panels A and C, respectively. *P < .01. (Asterisks indicating significance were withheld from panel B for visual clarity.)
Fetal and adult MK clusters were evaluated further by performing gene-expression analysis against a set of genes canonically associated with MKs at varying maturational stages (Figure 2B,D). Fetal lung MKs had the highest enrichment of several genes linked to MK development including Tubb1 and Gp1ba, markers of functional maturation66-68 (Figure 2B). From the adult populations, ABM-1 displayed the highest expression of MK-lineage genes, suggesting that this population represents the most mature cluster of BM-resident cells (Figure 2D). Notably, ABM-1 represents a very small fraction (4.5%) of the total MKs isolated from the BM. In contrast, the singular ALu exhibited significantly higher expression of mature MK markers, including Tubb1 and Gp1ba, compared with clusters ABM-2 and ABM-3. These less mature BM clusters showed significant enrichment for Gp1bb, a marker enriched in MK progenitor populations.69,70 Adult lung MKs also clustered more closely to ABM-1 (Figure 2C), suggesting a similar maturational profile that could be indicative of priming for platelet production. Collectively, these data suggest that there is a larger proportion of terminally mature MKs residing in the fetal and adult lung compared with the fetal liver and BM, respectively.
Fetal lung MKs display transcriptional priming for platelet and cytokine production
To further investigate the maturational development of lung MKs, fetal MK clusters were uploaded to the Klein Tools SPRING platform (https://kleintools.hms.harvard.edu/tools/spring).71 Visualization of fetal MK clusters with SPRING revealed a maturational trajectory of cells starting from progenitors and ending in terminally mature MKs (Figure 3A). This trajectory plot was annotated for directionality of maturation by assessing the cell cycle phase and the intensity of expression of key lineage markers that included Itga2b (CD41), Gp1ba (CD42b), and Tubb1 (supplemental Figure 4A-D). The intensity of expression of the progenitor markers Ptprc and Kit also helped to determine directionality of maturation (supplemental Figure 4E-F). Sixty-seven percent of the most mature clusters (Figure 3A, dashed box) were made up of fetal lung MKs. The top 150 genes enriched in the clusters of MKs outlined within this SPRING plot were cross-referenced with the WikiPathways mouse database. This analysis demonstrated an enrichment for cellular pathways important in proplatelet production, including ATP production, protein translation, and cytoskeletal rearrangement (Figure 3B). These cellular processes have been described to be more strongly associated with higher ploidy MKs.72 Collectively, this suggests that fetal lung MKs are in a more mature and metabolically active state that is primed for more efficient platelet production. Fetal MK clusters also displayed significant upregulation of various genes coding for cytokines and growth factors that are known to be packaged into platelet granules73-75 (Figure 3C). Notably, fetal lung MKs showed robust Tgfb1 expression and lung-specific enrichment for Igf1, both of which are critical in pulmonary development.30,76-78

Fetal lung MKs display transcriptional priming for platelet and cytokine production. (A) SPRING plot of fetal MK clusters. Directionality of maturation progresses from left to right and was determined based on key markers, as described in supplemental Figure 4. Percentages indicate which fraction of the outlined cells is represented by each respective cluster. (B) Gene set enrichment analysis of the corresponding clusters that are outlined in a dashed box in panel A. (C) Violin plots comparing the expression of various cytokines and growth factors across all fetal MK clusters. *P < .0001.
Fetal lung MKs display transcriptional priming for platelet and cytokine production. (A) SPRING plot of fetal MK clusters. Directionality of maturation progresses from left to right and was determined based on key markers, as described in supplemental Figure 4. Percentages indicate which fraction of the outlined cells is represented by each respective cluster. (B) Gene set enrichment analysis of the corresponding clusters that are outlined in a dashed box in panel A. (C) Violin plots comparing the expression of various cytokines and growth factors across all fetal MK clusters. *P < .0001.
Adult lung MKs exhibit higher ploidy and express markers of maturation
To complement these transcriptomic studies, adult MKs were also assessed via flow cytometry for ploidy and markers of maturity. Compared with BM-derived MKs, adult lung MKs possess a unique ploidy profile with a greater representation of high ploidy cells (Figure 4A; supplemental Figure 5). Furthermore, flow cytometry demonstrated that a significantly higher proportion of adult lung MKs expressed the key maturational marker CD42d (Figure 4B-C). Taken together with the scRNA-Seq analyses, these data demonstrate that the majority of adult lung MKs possess a more developmentally mature profile that may be indicative of their propensity for more efficient platelet production.
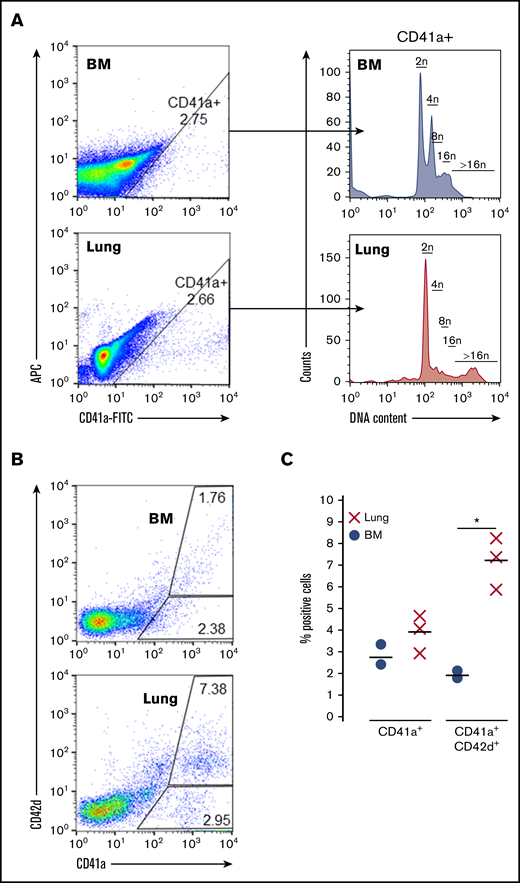
Adult lung MKs exhibit higher ploidy and express markers of maturation. (A) Ploidy analysis of CD41a+ MKs demonstrated a greater proportion of high ploidy MKs from the lung. (B-C) Flow cytometric assessment showed that a greater proportion of lung MKs was positive for the key maturation marker CD42d, which was significant across 3 biological replicates. BSA enrichment was used for these experiments. Ploidy analysis without BSA enrichment was also performed and showed similar results (supplemental Figure 5). "X" and "●" represent individual data points, whereas the horizontal bars are averages. *P < .05.
Adult lung MKs exhibit higher ploidy and express markers of maturation. (A) Ploidy analysis of CD41a+ MKs demonstrated a greater proportion of high ploidy MKs from the lung. (B-C) Flow cytometric assessment showed that a greater proportion of lung MKs was positive for the key maturation marker CD42d, which was significant across 3 biological replicates. BSA enrichment was used for these experiments. Ploidy analysis without BSA enrichment was also performed and showed similar results (supplemental Figure 5). "X" and "●" represent individual data points, whereas the horizontal bars are averages. *P < .05.
Lung MKs are skewed toward immunity and inflammation
Differential gene-expression analysis was performed on fetal and adult MK clusters, as defined by UMAP (Figures 5 and 6; full gene lists provided in supplemental Data 1 and 2). Using Enrichr analysis, the top 40 differentially expressed genes were cross referenced with the Gene Ontology Biological Process database. This analysis revealed robust enrichment for immune and inflammatory processes in fetal lung MKs (Figure 5). In contrast, fetal liver–derived populations were enriched for pathways related to vascular development and nuclear processes (supplemental Figure 6). Adult lung MKs also showed upregulation of immune and inflammatory processes (Figure 6). Alternatively, ABM-1 showed enrichment for processes important to proplatelet production, including cytoskeletal organization and protein localization to endosomes. The gene set enriched in ABM-1 was notably more comparable to ALu than to ABM-2 or ABM-3 (Figure 6, dashed box), further suggesting a more similar maturation profile between adult lung MKs and ABM-1, the most mature BM MK cluster. ABM-2 and ABM-3 demonstrated enrichment for pathways related to cell division and DNA processing, suggesting a more proliferative and immature profile (supplemental Figure 7). Differential gene-expression analyses comparing fetal lung MKs and adult lung MKs revealed the enrichment of major histocompatibility complex class II (MHCII) antigen-presentation genes specific to adult lung MKs (supplemental Figure 8; full gene lists provided in supplemental Data 3 and 4). To further assess the immune profile of lung MKs, gene-expression analysis of various surface immune markers and chemokines was performed (Figure 7A). Within this gene set, there was a distinct pattern of expression when comparing BM and lung MKs. Notably, lung MKs showed significantly higher transcriptional expression of many TLRs, chemokines, and the immune marker Cd74. To assess protein level expression, flow cytometric analyses of surface immune markers were performed on CD41a+/CD42d+ MKs (Figure 7B). Expression of TLR2, TLR4, and MHCII was significantly higher in lung MKs (Figure 7C), suggesting that lung MKs and the platelets that they produce may have significant roles in microbial surveillance and antigen presentation.
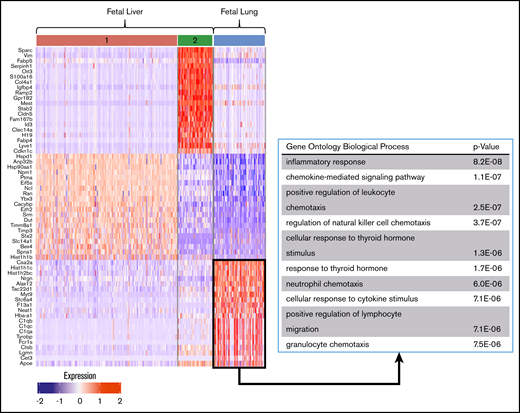
Differential gene-expression and pathway analysis demonstrate the immune phenotype of fetal lung MKs. Heat map and gene set enrichment analysis illustrating robust upregulation of immune/inflammatory genes in fetal lung MKs. The top 20 differentially expressed genes associated with each cluster are listed to the left of the heat map. The gene ontology pathways associated with the remaining clusters are shown in supplemental Figure 6.
Differential gene-expression and pathway analysis demonstrate the immune phenotype of fetal lung MKs. Heat map and gene set enrichment analysis illustrating robust upregulation of immune/inflammatory genes in fetal lung MKs. The top 20 differentially expressed genes associated with each cluster are listed to the left of the heat map. The gene ontology pathways associated with the remaining clusters are shown in supplemental Figure 6.
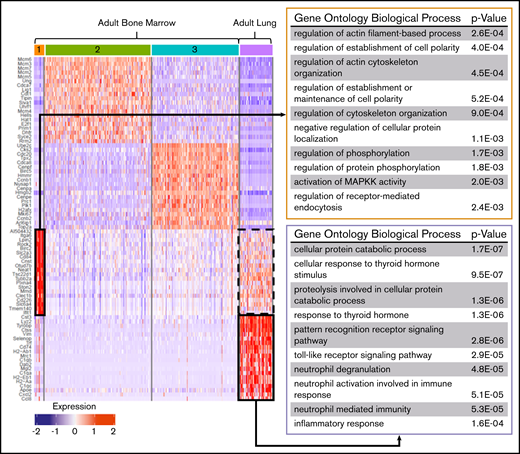
Differential gene expression and pathway analysis demonstrate the immune phenotype of adult lung MKs. Heat map and gene set enrichment analysis illustrating robust upregulation of immune/inflammatory genes in adult lung MKs. Adult BM cluster 1 showed enrichment for processes important in proplatelet production, and this gene set was notably also upregulated in the adult lung population (dashed black box). The top 20 differentially expressed genes associated with each cluster are listed to the left of the heat map. The gene ontology pathways associated with the remaining clusters are shown in supplemental Figure 7.
Differential gene expression and pathway analysis demonstrate the immune phenotype of adult lung MKs. Heat map and gene set enrichment analysis illustrating robust upregulation of immune/inflammatory genes in adult lung MKs. Adult BM cluster 1 showed enrichment for processes important in proplatelet production, and this gene set was notably also upregulated in the adult lung population (dashed black box). The top 20 differentially expressed genes associated with each cluster are listed to the left of the heat map. The gene ontology pathways associated with the remaining clusters are shown in supplemental Figure 7.
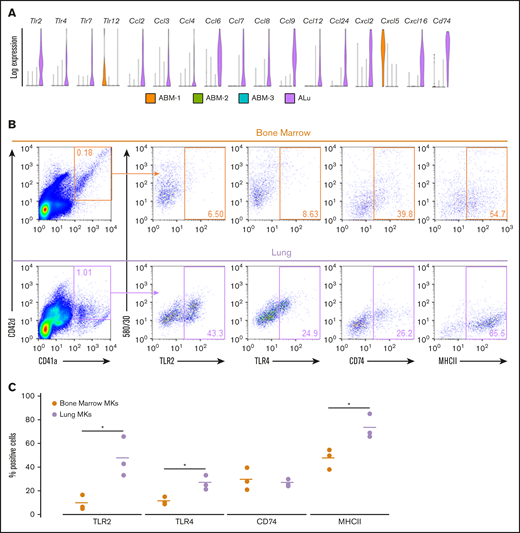
Adult Lung MKs exhibit significant upregulation of immune markers. (A) Gene-expression analysis of various TLRs, chemokines, and the surface immune marker Cd74 showed a distinct expression pattern with markedly more enrichment for many of these genes in lung MKs. (B) Flow cytometric assessment of TLR2, TLR4, CD74, and MHCII was performed on CD41a+/CD42d+ MKs. (C) Percentage of cells expressing these immune markers was quantified across 3 replicates, which demonstrated significantly higher expression of TLR2, TLR4, and MHCII in lung MKs. BSA enrichment was not used prior to immune marker analyses. *P < .05.
Adult Lung MKs exhibit significant upregulation of immune markers. (A) Gene-expression analysis of various TLRs, chemokines, and the surface immune marker Cd74 showed a distinct expression pattern with markedly more enrichment for many of these genes in lung MKs. (B) Flow cytometric assessment of TLR2, TLR4, CD74, and MHCII was performed on CD41a+/CD42d+ MKs. (C) Percentage of cells expressing these immune markers was quantified across 3 replicates, which demonstrated significantly higher expression of TLR2, TLR4, and MHCII in lung MKs. BSA enrichment was not used prior to immune marker analyses. *P < .05.
Discussion
In this study, we have explored the positioning of MKs within the lung architecture that may promote the specialization of hematopoietic and pulmonary tissue types. scRNA-Seq allowed us to profile individual MKs in syngeneic tissues, and it revealed a distinct transcriptional signature in lung MKs. Interestingly, lung MKs are more mature, may be primed for more efficient platelet production, and have an immune and inflammatory signature. Collectively, these findings suggest that the lung microenvironment plays a significant role in promoting the development of a lung-specific phenotype, further supporting the notion that the lung is a major site of MK development and functionalization.
Terminal maturation of MKs is a critical developmental process that increases an MK’s capacity to efficiently produce high volumes of platelets.79,80 During this process, MKs upregulate genes critical to the formation of proplatelets and their function in coagulation.80-83 Lung MKs exhibited significant upregulation of these key maturational genes, including Tubb1 and Gp1ba, both of which are strong indicators of late-stage maturity.66-68 Furthermore, pathway analysis showed enrichment for processes related to ATP production, protein synthesis, and cytoskeletal rearrangement, which are of relevance to the energy-intensive processes that preface platelet biogenesis.4,79 Other investigators have also demonstrated, via scRNA-Seq, that higher ploidy MKs are enriched for gene sets related to protein translation and cell metabolism,72 which aligns with the findings described above, as well as the high ploidy status of lung MKs that we have described here. Endomitotic divisions increase ploidy and cell size, which are critical for increasing protein production and expanding available cell membrane to fuel efficient platelet production.4,84 High platelet output is further supported by turbulent blood flow, which has been suggested to be a unique feature of the lung vasculature that further supports pulmonary platelet biogenesis.85 After budding off from their parent MK, platelets are carried off into the circulation to carry out a variety of functions.
One key function of platelets is the transport of cytokines and growth factors to support development and regeneration.49 Notably, other groups have demonstrated that lung-specific processes are supported by factors sourced from or enriched in platelets. Platelet-derived SDF1 promotes alveolar regeneration postpneumonectomy.31 Transforming growth factor-β1 is critical for the early embryonic patterning of healthy lungs.30,76,86-88 IGF1 is critical in the development of many tissues, including the lung.89-91 Although some of these studies highlight platelets as the source of these factors, the tissue source of the platelets is unclear. Despite differences in maturation, our analyses of fetal MK populations revealed robust transcriptional expression of Tgfb1 and lung-specific enrichment of Igf1. This suggests that the observed differences in expression of growth factors are not solely an artifact of the maturational state. Although the liver is the primary source of IGF1 during embryonic development, blood oxygenation is provided by the mother, and ∼16% of total blood flow passes through the murine pulmonary circulation.92 We also observed a low prevalence of platelets in fetal lung sections, suggesting that this reduced blood flow restricts the passage of platelets to the developing lung. As such, fetal lung MKs may be providing a locally enriched source of growth factors that support the development of the embryonic lung.
Recent investigation of MKs and platelets continues to reveal their previously underappreciated roles in inflammation and immunity.1-3,15,38 By using scRNA-Seq, we demonstrated that lung MKs in fetal and adult mice are indeed skewed toward an immune phenotype. Other investigators have purported that the adoption of an immune phenotype may be a result of interactions between the nonsterile environment of the lung niche and resident MKs.15 Although this may hold true in the adult lung, embryogenesis occurs under aseptic conditions, yet fetal lung MKs still develop an immune phenotype in the absence of lung microbiota.93,94 This suggests that the lung MK microenvironment promotes the adoption of an immune phenotype independently of influence from interactions with foreign antigens. The physiological significance of lung MKs being skewed toward a role in immunity remains unanswered. However, we demonstrate significant upregulation of TLR2, TLR4, and MHCII in lung MKs, which suggests that lung MKs and their platelet progeny may have important roles in microbial surveillance and antigen presentation. These findings were corroborated by a recent publication utilizing scRNA-Seq and functional immune studies to demonstrate the in vivo and ex vivo antigen-presentation capabilities specific to lung MKs.95 However, in contrast to our findings, this group demonstrated a lower ploidy profile in lung MKs. The discrepancy may be due, in part, to differences in the methodologies used for cell isolation and sorting strategies. Nevertheless, the collective findings from both studies highlight the need to further investigate the unique features of this cell population.
Our findings suggest the existence of symbiosis between the lung and resident MKs. In this scenario, the lung niche supports MK development and may further functionalize resident MKs. Reciprocally, resident MKs and platelets may support the lung in processes such as microbial defense and embryonic development. Further investigation into the development of lung MKs and their respective functional repertoire may shed light on the origin of platelets that promote or protect against the development of life-threatening lung disorders, such as lung fibrosis, acute respiratory distress syndrome, and tumor progression.28,96-100 This is particularly relevant in the wake of the SARS-COV-2 pandemic; patients presenting with severe and mild respiratory symptoms have exhibited life-threatening systemic coagulopathies, including stroke, multiorgan thrombosis, and disseminated intravascular coagulation.101-106 Furthermore, multiple groups have described an increase in alveolar MKs in histopathological lung sections from SARS-COV-2 patients.107,108 These findings, combined with the immune phenotype of lung MKs, raise questions about the involvement of lung MKs in the pathogenesis of SARS-COV-2 infection and other pulmonary diseases. Developing an enhanced understanding of lung MKs will further support the continuing search for novel therapeutics to treat lung diseases and will greatly advance our understanding of the organ-specific role of MKs.
Single-cell transcriptomic libraries have been deposited in the Gene Expression Omnibus database (accession number GSE152574; token: sdmtwiagzbsnfwl).
Data sharing requests should be sent to George J. Murphy (e-mail: gjmurphy@bu.edu).
Acknowledgments
The authors thank Brian R. Tilton and Anna Belkina (Boston University Flow Cytometry Core Facility), Yuriy Alekseyev, Ashley LeClerc, and M. J. Mistretta (Boston University Microarray and Sequencing Resource Core), and Michael T. Kirber (Boston University Cellular Imaging Core) for technical support, as well as Katya Ravid and Andrew (Larry) Frelinger for helpful discussions regarding experimental design.
This work was supported by a National Institutes of Health Training Grant for Hematology (5T32HL7501-36) and a National Institutes of Health Predoctoral National Research Service Award for MD/PhD Fellowships (1F30HL154552-01).
Authorship
Contribution: A.K.Y. and G.J.M. designed the experiments, collected, analyzed, and interpreted data, and prepared the manuscript; C.V.-M. performed bioinformatics/computational analysis and assisted with the experimental design and preparation of the manuscript; and S.H. and J.R.R. performed/supported data collection.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: George J. Murphy, Department of Medicine, Boston University School of Medicine, 670 Albany St, 2nd Floor, Boston, MA 02118-2393; e-mail: gjmurphy@bu.edu.

