

Key Points
Development of FVIII inhibitors within the first 50 EDs to FVIII is associated with distinct antibody signatures.
Patients with persistent FVIII inhibitors develop unique signatures of FVIII-binding IgG1, followed by IgG3 and IgG4.

Abstract
Preventing factor VIII (FVIII) inhibitors following replacement therapies with FVIII products in patients with hemophilia A remains an unmet medical need. Better understanding of the early events of evolving FVIII inhibitors is essential for risk identification and the design of novel strategies to prevent inhibitor development. The Hemophilia Inhibitor Previously Untreated Patients (PUPs) Study (HIPS; www.clinicaltrials.gov #NCT01652027) is the first prospective cohort study to evaluate comprehensive changes in the immune system during the first 50 exposure days (EDs) to FVIII in patients with severe hemophilia A. HIPS participants were enrolled prior to their first exposure to FVIII or blood products (“true PUPs”) and were evaluated for different immunological and clinical parameters at specified time points during their first 50 EDs to a single source of recombinant FVIII. Longitudinal antibody data resulting from this study indicate that there are 4 subgroups of patients expressing distinct signatures of FVIII-binding antibodies. Subgroup 1 did not develop any detectable FVIII-binding immunoglobulin G (IgG) antibodies. Subgroup 2 developed nonneutralizing, FVIII-binding IgG1 antibodies, but other FVIII-binding IgG subclasses were not observed. Subgroup 3 developed transient FVIII inhibitors associated with FVIII-binding IgG1 antibodies, similar to subgroup 2. Subgroup 4 developed persistent FVIII inhibitors associated with an initial development of high-affinity, FVIII-binding IgG1 antibodies, followed by IgG3 and IgG4 antibodies. Appearance of FVIII-binding IgG3 was always associated with persistent FVIII inhibitors and the subsequent development of FVIII-binding IgG4. Some of the antibody signatures identified in HIPS could serve as candidates for early biomarkers of FVIII inhibitor development.
Introduction
Hemophilia A is a congenital bleeding disorder caused by a deficiency in biologically active factor VIII (FVIII). Most patients receive FVIII concentrates throughout the course of replacement therapies. These therapies can be complicated by the development of neutralizing antibodies directed against FVIII (FVIII inhibitors), which are usually observed after 5 to 15 exposure days (EDs) to exogenous FVIII in ∼30% of previously untreated patients (PUPs) with severe hemophilia A.1-4 Morbidity and cost of care for patients with FVIII inhibitors are substantially increased which is why novel treatment strategies to prevent FVIII inhibitor development in patients undergoing FVIII replacement therapies are required.5-7
FVIII inhibitors are quantified by their ability to inhibit FVIII activity using the Bethesda assay or the Nijmegen-modified Bethesda assay.8,9 Recent studies indicated that FVIII inhibitors represent only a fraction of the potential antibody response directed against FVIII.10-12 Using a combination of Bethesda assays and sensitive enzyme-linked immunosorbent assays (ELISAs), circulating antibodies directed against FVIII were detected in patients with and without FVIII inhibitors, indicating the development of both, neutralizing and nonneutralizing antibodies. Moreover, nonneutralizing antibodies against FVIII were also found in some healthy individuals.10,11
Why some patients develop FVIII inhibitors and others do not is poorly understood. There is evidence that both genetic risk factors (eg, F8 mutations, family history of FVIII inhibitors, polymorphisms in genes encoding immune regulatory proteins, and ethnicity backgrounds) and nongenetic risk factors (eg, treatment-related and environmental factors) influence the development of these antibodies.13-17 However, the molecular mechanisms responsible for the initiation or prevention of FVIII inhibitors in patients have not been well explained. Hofbauer et al reported that antibodies detected in patients with FVIII inhibitors are predominantly of high affinity, whereas antibodies found in some patients without FVIII inhibitors and in some healthy individuals are predominantly of medium or low affinity.11 These data support previous findings in hemophilic mouse models indicating that the development of neutralizing antibodies directed against FVIII depends on cognate interactions between FVIII-specific B cells and FVIII-specific T cells, for example follicular helper T cells, in specific structures of secondary lymphoid organs called germinal centers.18,19 These cognate interactions drive affinity maturation and class-switch recombination of antibodies as well as the differentiation of B cells into memory B cells and long-lived plasma cells secreting high-affinity antibodies.20,21 On the other hand, antibodies with low and medium affinity could be the result of extrafollicular B-cell differentiation, which can be either dependent or independent of their cognate interaction with antigen-specific T cells.20
To further explain the underlying immune mechanisms that are responsible for the generation of high- and low-affinity antibodies against FVIII in patients, a good understanding of the longitudinal events associated with these different antibody responses and the potential interdependence of nonneutralizing and neutralizing antibody responses is required. Therefore, prospective, longitudinal clinical studies monitoring the development and characteristic features of FVIII-binding antibodies as well as underlying immune mechanisms in PUPs, starting prior to the first dose of FVIII, are important. Such studies could not only facilitate early risk identification for FVIII inhibitor development in individual patients but also support the design of novel treatment approaches to prevent FVIII inhibitor development.
The Hemophilia Inhibitor PUPs Study (HIPS; www.clinicaltrials.gov #NCT01652027) is a prospective, longitudinal, observational study that was designed to identify early biomarkers of FVIII inhibitor development and prospectively evaluate early changes in the immune system upon exposure to FVIII in patients with severe hemophilia A. HIPS patients were enrolled prior to their first exposure to FVIII or blood products (ie, true PUPs) and were evaluated at specified time points during their first 50 EDs to a single source of full-length, standard half-life, recombinant FVIII concentrate. In this article, we present data indicating different subpopulations of patients who express distinct signatures of longitudinal antibody responses directed against FVIII.
Materials and methods
Study design
HIPS is a multicenter, investigator-initiated, prospective longitudinal cohort study of previously untreated infants with severe hemophilia A (www.clinicaltrials.gov #NCT01652027). Sixteen US and European hemophilia treatment centers (supplemental Table 1) participated in HIPS and performed HIPS qualification testing to ensure quality of the biological specimens. Each site obtained approval from their institutional review board or independent ethics committee.
Enrollment criteria, treatment, and clinical procedures
Eligible patients were previously untreated infants with a baseline FVIII coagulant activity (FVIII:C) <0.01 IU/mL confirmed in the central laboratory at the Medical University of Vienna. Minimal weight of 3.5 kg for patients at enrollment was required to allow for 7.5 mL of blood to be drawn at each visit with minimal risk to the patient. Patients were excluded from HIPS if they had prior exposure to clotting factor concentrates or blood products, including packed red blood cells, platelets, plasma, or cryoprecipitate. Written informed consent, approved by the appropriate institutional review board or independent ethics committee, was obtained from a parent or legally authorized representative of each study participant.
Patients were treated with a recombinant FVIII concentrate (antihemophilic factor [recombinant], Advate; Baxalta US, a Takeda company, Lexington, MA) as their only source of FVIII concentrate for prophylaxis, treatment of bleeding episodes, surgical procedures, and immune tolerance induction, if required. The type of regimen (prophylaxis vs on demand), dose, and frequency of infusions were at the discretion of the investigator. Patients were followed for 50 EDs or 3 years, whichever came first.
Screening and baseline evaluations took place prior to the first FVIII infusion. Patients underwent clinical evaluation visits every 12 weeks (±10 days) after the first ED during which the medical, social, and family histories were updated and a physical examination was performed. Parents were required to maintain a paper diary to record information about FVIII infusions, including date of infusion, number of units infused, number of vials used, lot number(s), reason for infusion, and any adverse events. In addition, the diary included information about infections, immunizations, medications and dietary supplements, surgeries, and trauma.
Blood sample schedule
Blood samples were drawn at baseline before FVIII exposure and then 7 days (+2 days) after the first ED. Subsequently, samples were taken 5 days (±2 days) after the fifth, 10th, 20th, 30th, 40th, and 50th ED for the same set of measurements as indicated in supplemental Figure 1. In the event that not all samples for the first, fifth, 10th, 20th, 30th, 40th, or 50th post-FVIII exposure visit could be obtained, a scheduled blood draw was performed after the subsequent FVIII exposure following the same visit window of 5 days (±2 days) after exposure to FVIII. Further details for the blood sampling schedule are provided in supplemental Materials and methods. Blood volumes required for analytical procedures during the HIPS study are shown in Table 1.
Blood volumes required for analytical procedures during the HIPS study
| Blood sample analytics | Sampled blood volume, mL* |
|---|---|
| Screening and baseline samples | |
| FVIII activity in plasma | 1.8 |
| Transcriptome profiling in PBMCs after short-term in vitro stimulation with FVIII; FVIII-specific antibody analytics | 4.0 |
| Epigenetic immune cell counting; whole-blood transcriptome profiling | 2.0 |
| Post-FVIII exposure samples | |
| FVIII inhibitor testing; whole-genome profiling (once); FVIII mutation analysis (once) | 2.7 |
| Transcriptome profiling in PBMCs after short-term in vitro stimulation with FVIII; FVIII-specific antibody analytics | 4.0 |
| Epigenetic immune cell counting; whole-blood transcriptome profiling | 2.0 |
| Blood sample analytics | Sampled blood volume, mL* |
|---|---|
| Screening and baseline samples | |
| FVIII activity in plasma | 1.8 |
| Transcriptome profiling in PBMCs after short-term in vitro stimulation with FVIII; FVIII-specific antibody analytics | 4.0 |
| Epigenetic immune cell counting; whole-blood transcriptome profiling | 2.0 |
| Post-FVIII exposure samples | |
| FVIII inhibitor testing; whole-genome profiling (once); FVIII mutation analysis (once) | 2.7 |
| Transcriptome profiling in PBMCs after short-term in vitro stimulation with FVIII; FVIII-specific antibody analytics | 4.0 |
| Epigenetic immune cell counting; whole-blood transcriptome profiling | 2.0 |
PBMC, peripheral blood mononuclear cell.
The total blood volume sampled did not exceed 8 mL/kg body weight at any month during the study
F8 genotyping
F8 genotyping was performed at BloodWorks Northwest, Seattle, WA. Details are provided in supplemental Materials and methods.
Detection of neutralizing antibodies
FVIII inhibitor analysis was performed in the central laboratory at the Medical University of Vienna using a Nijmegen-modified Bethesda assay. The lower limit of inhibitor detection was 0.4 Bethesda (BU)/mL. A patient was considered FVIII inhibitor positive if neutralizing antibodies were detected at ≥0.6 BU/mL in at least 2 consecutive exposure samples. A positive inhibitor that peaked at ≤5 BU/mL after rechallenge with FVIII was considered a low-titer inhibitor, and a positive inhibitor >5 BU/mL was considered a high-titer inhibitor. Transient inhibitors were defined as those that were of low titer (≤5 BU/mL) that disappeared within 6 months and the patient was able to remain on FVIII therapy for the treatment of hemorrhages.22
Detection of FVIII-binding antibodies
FVIII-specific antibodies of immunoglobulin isotypes immunoglobulin A (IgA), IgM, and IgG subclasses 1 to 4 were analyzed using a fully validated ELISA platform as described elsewhere.10,11
Assessment of apparent affinities of FVIII-binding antibodies
The apparent affinities of FVIII-binding antibodies for IgA and IgG subclasses were assessed using a competition-based ELISA approach. The test principle, technical details, and validation of the affinity ELISA platform were described by Hofbauer et al.11 In brief, the competition-based ELISA approach provides apparent affinities in equilibrium; it analyzes free antibodies against FVIII present in diluted human plasma after preincubation of the antibody solution with preselected molar concentrations of FVIII. Based on the assumption of equimolar interaction between antibodies and FVIII antigen, this approach enables apparent affinity determination without antibody purification. The assessment of affinity constants is based on the availability of antibody for binding to FVIII-coated ELISA plates after competition with FVIII in solution. Data for apparent affinity constants (KA [M−1]) were derived from nonlinear regression modeling of competition ELISA δ optical densities as described by Stevens et al23 and Hofbauer et al.11 The calculations of apparent affinity constants and the best-fit model for affinity cluster analyses were done as described previously.11
Statistical analyses
In order to compare antibody signatures in patients with FVIII inhibitors (subgroup 4; Table 2) and in patients with nonneutralizing antibodies (subgroup 2; Table 2), medians and interquartile ranges (IQRs) for titers and apparent affinity constants of FVIII-binding antibodies, differentiated for IgG subclasses, IgM and IgA, were calculated. The calculations for the IQR included all antibody data for each patient in the respective subgroup at each time point analyzed. Group comparisons were done using GraphPad Prism 8.0 and IBM SPSS Statistics version 23 (IBM). Medians, 95% confidence intervals (CIs), and IQRs were used to describe data. Comparisons between the groups were done using the Mann-Whitney U test. For all analyses, a value of P < .05 was considered as statistically significant.
Patient characteristics stratified in 4 subgroups with distinct signatures of FVIII-binding antibodies developed during the HIPS study
| Patient no. | F8 mutation | Age at first FVIII infusion, mo | Peak FVIII inhibitor titer, BU/mL | First FVIII inhibitor detection | Ethnic background | Family history of hemophilia A | Family history of FVIII inhibitors |
|---|---|---|---|---|---|---|---|
| Subgroup 1: no FVIII-binding IgG antibodies | |||||||
| 1 | Missense | 9.1 | <0.4 | NA | White | No | No |
| 2 | Intron 22 inversion | 12.7 | <0.4 | NA | White | Yes | Yes |
| 3 | Missense | 2.8 | <0.4 | NA | White | Yes | No |
| 4 | Missense | 2.5 | <0.4 | NA | Black | Yes | No |
| 5 | Duplication | 7.8 | <0.4 | NA | White | No | No |
| 6 | Frameshift | 16.6 | <0.4 | NA | White | Yes | Yes |
| 7 | Nonsense | 6.7 | <0.4 | NA | White | Yes | Unknown |
| Subgroup 2: non-neutralizing FVIII-binding antibodies | |||||||
| 8 | Intron 22 inversion | 9.3 | <0.4 | NA | White | No | No |
| 9 | Missense | 19.9 | <0.4 | NA | Black | Yes | Yes |
| 10 | Intron 22 inversion | 3.6 | <0.4 | NA | White | No | No |
| 11 | Missense | 13.5 | <0.4 | NA | White | No | No |
| 12 | Intron 1 inversion | 12.0 | <0.4 | NA | White | No | No |
| 13 | Missense | 10.7 | <0.4 | NA | White | No | No |
| 14 | Intron 22 inversion | 0.4 | <0.4 | NA | White | Yes | Yes |
| Subgroup 3: transient FVIII inhibitors | |||||||
| 15 | Frameshift | 14.7 | 1.8 | ED20 | Black | Yes | Yes |
| 16 | Intron 22 inversion | 9.7 | 1.4 | ED12 | White | Yes | No |
| Subgroup 4: persistent FVIII inhibitors | |||||||
| 17 | Large deletion | 10.2 | 4.5 | ED20 | Hispanic | No | No |
| 18 | Large deletion | 12.0 | 7 | ED6 | Hispanic | Yes | Unknown |
| 19 | Intron 22 inversion | 12.6 | 796 | ED10 | White | Yes | Unknown |
| 20 | Duplication | 12.0 | 9.5 | ED10 | Hispanic | Yes | Yes |
| 21 | Intron 22 inversion | 10.3 | 323 | ED10 | White | Yes | Unknown |
| 22 | Intron 22 inversion | 6.3 | 3049 | ED10 | Asian or Pacific | Yes | No |
| 23 | Intron 22 inversion | 9.2 | 124 | ED11 | White | Yes | Yes |
| Patient no. | F8 mutation | Age at first FVIII infusion, mo | Peak FVIII inhibitor titer, BU/mL | First FVIII inhibitor detection | Ethnic background | Family history of hemophilia A | Family history of FVIII inhibitors |
|---|---|---|---|---|---|---|---|
| Subgroup 1: no FVIII-binding IgG antibodies | |||||||
| 1 | Missense | 9.1 | <0.4 | NA | White | No | No |
| 2 | Intron 22 inversion | 12.7 | <0.4 | NA | White | Yes | Yes |
| 3 | Missense | 2.8 | <0.4 | NA | White | Yes | No |
| 4 | Missense | 2.5 | <0.4 | NA | Black | Yes | No |
| 5 | Duplication | 7.8 | <0.4 | NA | White | No | No |
| 6 | Frameshift | 16.6 | <0.4 | NA | White | Yes | Yes |
| 7 | Nonsense | 6.7 | <0.4 | NA | White | Yes | Unknown |
| Subgroup 2: non-neutralizing FVIII-binding antibodies | |||||||
| 8 | Intron 22 inversion | 9.3 | <0.4 | NA | White | No | No |
| 9 | Missense | 19.9 | <0.4 | NA | Black | Yes | Yes |
| 10 | Intron 22 inversion | 3.6 | <0.4 | NA | White | No | No |
| 11 | Missense | 13.5 | <0.4 | NA | White | No | No |
| 12 | Intron 1 inversion | 12.0 | <0.4 | NA | White | No | No |
| 13 | Missense | 10.7 | <0.4 | NA | White | No | No |
| 14 | Intron 22 inversion | 0.4 | <0.4 | NA | White | Yes | Yes |
| Subgroup 3: transient FVIII inhibitors | |||||||
| 15 | Frameshift | 14.7 | 1.8 | ED20 | Black | Yes | Yes |
| 16 | Intron 22 inversion | 9.7 | 1.4 | ED12 | White | Yes | No |
| Subgroup 4: persistent FVIII inhibitors | |||||||
| 17 | Large deletion | 10.2 | 4.5 | ED20 | Hispanic | No | No |
| 18 | Large deletion | 12.0 | 7 | ED6 | Hispanic | Yes | Unknown |
| 19 | Intron 22 inversion | 12.6 | 796 | ED10 | White | Yes | Unknown |
| 20 | Duplication | 12.0 | 9.5 | ED10 | Hispanic | Yes | Yes |
| 21 | Intron 22 inversion | 10.3 | 323 | ED10 | White | Yes | Unknown |
| 22 | Intron 22 inversion | 6.3 | 3049 | ED10 | Asian or Pacific | Yes | No |
| 23 | Intron 22 inversion | 9.2 | 124 | ED11 | White | Yes | Yes |
NA, not applicable.
Results
Screening, enrollment, and baseline characteristics of patients
Twenty-six patients were screened and 25 determined to be eligible (1 subject with FVIII >1% at central laboratory was excluded) for the study. Of these 25 patients enrolled, 1 patient was withdrawn after ED1 due to the lack of compliance, and 1 patient withdrew after ED10 due to investigator choice (change in treatment), leaving 23 patients who completed 50 EDs. Baseline characteristics of these 23 subjects, such as age at first FVIII infusion (0.4-16.6 months), ethnical background (16 White, 3 Black, 3 Hispanic, and 1 Asian/Pacific), family history of hemophilia A and of FVIII inhibitors, hospitalizations during the study, infections, and FVIII treatment regimens are shown in Tables 2 and 3. A total of 17 patients (73.9%) carried high-risk or moderate-risk F8 mutations (intron 22 inversion, 9; duplications, 2; frameshift, 2; large deletions, 2; intron 1 inversion, 1; and nonsense, 1), while 6 patients (26.1%) had low-risk mutations (missense).
Hospitalizations and FVIII treatment regimens for HIPS subjects, stratified in 4 subgroups with distinct signatures of FVIII-binding antibodies developed during the HIPS study
| Patient no. | Hospitalizations during study | EDs when prophylaxis and ITI (if applicable) were initiated (FVIII dose in IU/kg and treatment frequency) |
|---|---|---|
| Subgroup 1: no FVIII-binding IgG antibodies | ||
| 1 | No hospitalizations reported | ED1 (55 qw, 55 biw, 75 biw) |
| 2 | 4 hospitalizations: tongue bleed, head trauma, muscle bleed, port implant | ED35 (25 qw, 25 biw) |
| 3 | 1 hospitalization: port implant and circumcision | ED13 (25 biw & 50 qw; 50 biw & 70 qw) |
| 4 | No hospitalizations reported | ED5 (50 biw, 45 biw) |
| 5 | No hospitalizations reported | ED5 (90 qw, 55qw, 65 qw, 60 biw) |
| 6 | No hospitalizations reported | ED1 (70 qw, 95 qw, 60 qw, 60 biw) |
| 7 | No hospitalizations reported | ED1 (70 qw, 45 qw, 20 biw, 20 tiw) |
| Subgroup 2: nonneutralizing FVIII-binding antibodies | ||
| 8 | 1 hospitalization: port implant | ED1 (25 biw, 25 tiw) |
| 9 | No hospitalizations reported | ED3 (55 qw, 55 biw) |
| 10 | 3 hospitalizations: muscle bleed, left knee joint bleed, port implant | ED7 (25 – 55 qw, 25 biw, 20 tiw) |
| 11 | No hospitalizations reported | ED17 (25 tiw) |
| 12 | 1 hospitalization: muscle bleed (back) | ED1 (50 qw, 60 qw, 35 biw) |
| 13 | No hospitalizations reported | ED6 (30 qw, 30 biw) |
| 14 | No hospitalizations reported | ED8 (40 qw, 50 qw, 50 biw) |
| Subgroup 3: transient FVIII inhibitors | ||
| 15 | No hospitalizations reported | ED6 (60 qw, 45 qw, 50 qw) |
| 16 | 1 hospitalization: port implant (procedure occurred 3 mo after first inhibitor detection) | ED3 (90 qw, 55 qw, 65 biw) |
| Subgroup 4: persistent FVIII inhibitors | ||
| 17 | 1 hospitalization: port implant (procedure occurred 20 d prior to inhibitor detection) | ED1 (55 qw); ITI 225 qd (initiated on ED12) |
| 18 | No hospitalizations reported | ED1 (45 - 60 qw); ITI 210 qd (initiated on ED7) |
| 19 | 2 hospitalizations: intraspinal bleed and port implant, MRI and diagnosis after intraspinal bleed (all events occurred 1.5 y after inhibitor detection) | ED2 (35 qw); ITI 105 qd (initiated on ED13) |
| 20 | 1 hospitalization: port implant (procedure occurred 2 wk after inhibitor detection) | ED2 (120 qw); ITI 560 qd (initiated on ED16) |
| 21 | 3 hospitalizations: port implant (3 d prior to inhibitor detection), 2 infections (1 wk after inhibitor detection) | ED1 (30 qw); ITI 190 qd (initiated on ED11) |
| 22 | 3 hospitalizations: 2 fistulas, 1 hematoma episode (all events occurred after inhibitor detection) | ED1: 35 biw, ITI 210 qd (initiated on ED12) |
| 23 | 1 hospitalization: shunt for venous access (procedure occurred 2 mo after inhibitor detection) | ED3: 30 qw, ITI 155 qd (initiated on ED13) |
| Patient no. | Hospitalizations during study | EDs when prophylaxis and ITI (if applicable) were initiated (FVIII dose in IU/kg and treatment frequency) |
|---|---|---|
| Subgroup 1: no FVIII-binding IgG antibodies | ||
| 1 | No hospitalizations reported | ED1 (55 qw, 55 biw, 75 biw) |
| 2 | 4 hospitalizations: tongue bleed, head trauma, muscle bleed, port implant | ED35 (25 qw, 25 biw) |
| 3 | 1 hospitalization: port implant and circumcision | ED13 (25 biw & 50 qw; 50 biw & 70 qw) |
| 4 | No hospitalizations reported | ED5 (50 biw, 45 biw) |
| 5 | No hospitalizations reported | ED5 (90 qw, 55qw, 65 qw, 60 biw) |
| 6 | No hospitalizations reported | ED1 (70 qw, 95 qw, 60 qw, 60 biw) |
| 7 | No hospitalizations reported | ED1 (70 qw, 45 qw, 20 biw, 20 tiw) |
| Subgroup 2: nonneutralizing FVIII-binding antibodies | ||
| 8 | 1 hospitalization: port implant | ED1 (25 biw, 25 tiw) |
| 9 | No hospitalizations reported | ED3 (55 qw, 55 biw) |
| 10 | 3 hospitalizations: muscle bleed, left knee joint bleed, port implant | ED7 (25 – 55 qw, 25 biw, 20 tiw) |
| 11 | No hospitalizations reported | ED17 (25 tiw) |
| 12 | 1 hospitalization: muscle bleed (back) | ED1 (50 qw, 60 qw, 35 biw) |
| 13 | No hospitalizations reported | ED6 (30 qw, 30 biw) |
| 14 | No hospitalizations reported | ED8 (40 qw, 50 qw, 50 biw) |
| Subgroup 3: transient FVIII inhibitors | ||
| 15 | No hospitalizations reported | ED6 (60 qw, 45 qw, 50 qw) |
| 16 | 1 hospitalization: port implant (procedure occurred 3 mo after first inhibitor detection) | ED3 (90 qw, 55 qw, 65 biw) |
| Subgroup 4: persistent FVIII inhibitors | ||
| 17 | 1 hospitalization: port implant (procedure occurred 20 d prior to inhibitor detection) | ED1 (55 qw); ITI 225 qd (initiated on ED12) |
| 18 | No hospitalizations reported | ED1 (45 - 60 qw); ITI 210 qd (initiated on ED7) |
| 19 | 2 hospitalizations: intraspinal bleed and port implant, MRI and diagnosis after intraspinal bleed (all events occurred 1.5 y after inhibitor detection) | ED2 (35 qw); ITI 105 qd (initiated on ED13) |
| 20 | 1 hospitalization: port implant (procedure occurred 2 wk after inhibitor detection) | ED2 (120 qw); ITI 560 qd (initiated on ED16) |
| 21 | 3 hospitalizations: port implant (3 d prior to inhibitor detection), 2 infections (1 wk after inhibitor detection) | ED1 (30 qw); ITI 190 qd (initiated on ED11) |
| 22 | 3 hospitalizations: 2 fistulas, 1 hematoma episode (all events occurred after inhibitor detection) | ED1: 35 biw, ITI 210 qd (initiated on ED12) |
| 23 | 1 hospitalization: shunt for venous access (procedure occurred 2 mo after inhibitor detection) | ED3: 30 qw, ITI 155 qd (initiated on ED13) |
The last column, “EDs when prophylaxis and ITI (if applicable) were initiated,” includes the dose and frequency of prophylaxis and any changes to dose and frequency. Dose was calculated using the most recent prior weight measured (at screening or interval visit) and the FVIII dose in IU/kg reported on the infusion diary. All subjects received prophylaxis at some stage during the HIPS study. Subjects who did not start prophylaxis on ED1 were treated on demand until the exposure day specified in the table.
biw, twice a week; ITI, immune tolerance induction; MRI, magnetic resonance imaging; qd, daily; qw, once a week; tiw, 3 times a week.
Incidence of FVIII inhibitors
Nine of the 23 patients who completed 50 EDs developed FVIII inhibitors (6 high titer and 3 low titer). Two of the low-titer inhibitors were transient (Table 2). All 7 patients who developed persistent FVIII inhibitors (6 high titer and 1 low titer) carried high-risk or moderate-risk F8 mutations (Table 2).
Longitudinal monitoring for FVIII-binding antibodies
The analyses of FVIII-binding antibodies revealed 4 subgroups of patients with distinct antibody signatures: subgroup 1 (no FVIII-binding IgG antibodies), subgroup 2 (nonneutralizing FVIII-binding antibodies), subgroup 3 (transient FVIII inhibitors), and subgroup 4 (persistent FVIII inhibitors) (Table 2).
Subgroup 1: Seven patients did not develop inhibitors and did not have detectable FVIII-binding IgG throughout the course of the study (subgroup 1 in Tables 2 and 3; Figure 1; supplemental Figure 2). The transient appearance of low-titer FVIII-binding IgM or IgA antibodies at single EDs was observed in 2 of the 7 patients. One nonsense mutation, 1 intron 22 inversion, 3 missense mutations, 1 duplication, and 1 frameshift mutation of the F8 gene were found in these patients (Table 2).
Subgroup 2: Seven patients who were negative for FVIII inhibitors throughout the observation period developed FVIII-binding IgG1 (subgroup 2 in Tables 2 and 3; Figure 2; supplemental Figure 3). The IgG1 antibodies were transient in 3 patients and persisted to the end of the observation period in 4 patients. The apparent affinity constants of these antibodies revealed 1 or 2 affinity clusters (Figure 2; supplemental Figure 3). The kinetics of FVIII-binding IgG1 and their persistence and apparent affinity constants showed patient-specific characteristics. For example, patient 9 had detectable FVIII-binding IgG1 antibodies as early as ED5, but the antibodies were transient in nature. In contrast, FVIII-binding IgG1 antibodies observed in patient 14 were first detected at ED5 but persisted throughout the study observation period (Figure 2). FVIII-binding IgM was not observed in any patient of this subgroup, and low-titer transient FVIII-binding IgA was observed in 2 patients of this subgroup. Three missense mutations, 3 intron 22 inversions, and 1 intron 1 inversion of the F8 gene were found in patients of subgroup 2 (Table 2).
Subgroup 3: Two patients developed FVIII-binding IgG1 and low-titer transient FVIII inhibitors (peak titers ≤1.8). The apparent affinity constants of the IgG1 antibodies revealed 2 affinity clusters (subgroup 3 in Tables 2 and 3; Figure 3). Neither of these 2 patients developed any other FVIII-binding IgG subclass antibody. FVIII-binding IgM or IgA antibodies were not observed in these patients. One intron 22 inversion and 1 frameshift mutation of the F8 gene were found in these patients (Table 2).
Subgroup 4: The remaining 7 patients developed persistent FVIII inhibitors with peak titers ranging between 4.5 and 3049 BU/mL (subgroup 4 in Tables 2 and 3; Figure 4; supplemental Figure 4). FVIII inhibitors were first detected within 20 EDs (EDs 6-20) and persisted until the end of the study observation period. All patients in subgroup 4 initially developed high-affinity FVIII-binding IgG1 antibodies, followed by FVIII-binding IgG3 and IgG4. FVIII-binding IgG3 was usually observed subsequent to FVIII-specific IgG1. FVIII-specific IgG4 appeared later than IgG1 and either at the same time or subsequent to IgG3. FVIII-specific IgG2, observed in 3 patients only, was first detected subsequent to IgG1 and either subsequent to IgG3 and IgG4 or at the same time as IgG3 and IgG4. The highest apparent affinity constants were observed for FVIII-binding IgG4. FVIII-binding IgM and IgA antibodies of low titer were found in 1 of the 7 patients. Two large deletions, 4 intron 22 inversions, and 1 duplication in the F8 gene were found in patients of subgroup 4 (Table 2).
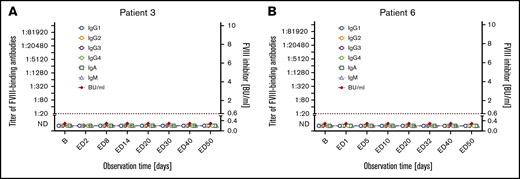
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in 2 representative examples of patients in subgroup 1. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, IgM as indicated) and FVIII inhibitors (BU/mL) for 2 representative examples of patients in subgroup 1, patient 3 (A) and patient 6 (B), who did not develop FVIII inhibitors throughout the study period. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). The data for the remaining 5 patients of subgroup 1 are shown in supplemental Figure 2. B, baseline; ND, not detectable (below the detection limit of 1:20 for FVIII-binding antibodies).
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in 2 representative examples of patients in subgroup 1. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, IgM as indicated) and FVIII inhibitors (BU/mL) for 2 representative examples of patients in subgroup 1, patient 3 (A) and patient 6 (B), who did not develop FVIII inhibitors throughout the study period. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). The data for the remaining 5 patients of subgroup 1 are shown in supplemental Figure 2. B, baseline; ND, not detectable (below the detection limit of 1:20 for FVIII-binding antibodies).
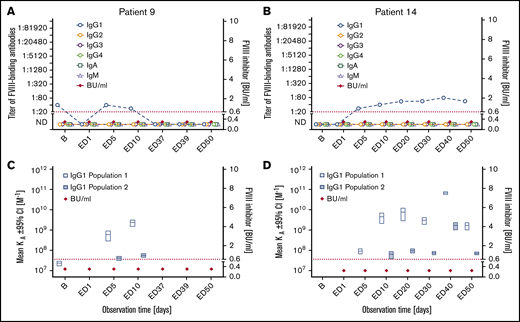
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in 2 representative examples of patients in subgroup 2. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, and IgM, as indicated) and FVIII inhibitors (BU/mL) for 2 representative examples of patients in subgroup 2, patient 9 (A) and patient 14 (B), who developed FVIII-binding IgG1 antibodies but did not develop FVIII inhibitors throughout the study period. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). (C-D) Apparent affinity constants of FVIII-binding IgG1 antibodies (mean KA) and FVIII inhibitors (BU/mL) in patient 9 (C) and patient 14 (D). Data for apparent affinity constants include the 95% CIs for up to 2 IgG1 affinity clusters (open blue bars, IgG1 population 1; closed blue bars, IgG1 population 2). The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). KA, apparent affinity constant. The data for the remaining 5 patients of subgroup 2 are shown in supplemental Figure 3.
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in 2 representative examples of patients in subgroup 2. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, and IgM, as indicated) and FVIII inhibitors (BU/mL) for 2 representative examples of patients in subgroup 2, patient 9 (A) and patient 14 (B), who developed FVIII-binding IgG1 antibodies but did not develop FVIII inhibitors throughout the study period. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). (C-D) Apparent affinity constants of FVIII-binding IgG1 antibodies (mean KA) and FVIII inhibitors (BU/mL) in patient 9 (C) and patient 14 (D). Data for apparent affinity constants include the 95% CIs for up to 2 IgG1 affinity clusters (open blue bars, IgG1 population 1; closed blue bars, IgG1 population 2). The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). KA, apparent affinity constant. The data for the remaining 5 patients of subgroup 2 are shown in supplemental Figure 3.
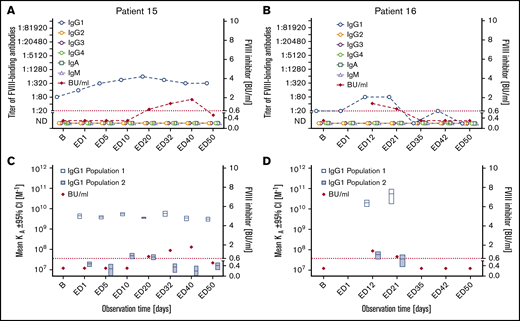
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in the 2 patients in subgroup 3. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, and IgM, as indicated) and FVIII inhibitors (BU/mL) for the 2 patients in subgroup 3, patient 15 (A) and patient 16 (B), who developed FVIII-binding IgG1 antibodies and transient low-titer FVIII inhibitors. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). (C-D) Apparent affinity constants of FVIII-binding IgG1 antibodies (mean KA) and FVIII inhibitors (BU/mL) in patient 15 (C) and patient 16 (D). Data for apparent affinity constants include the 95% CIs for up to 2 IgG1 affinity clusters (open blue bars, IgG1 population 1; closed blue bars, IgG1 population 2). The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL).
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in the 2 patients in subgroup 3. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, and IgM, as indicated) and FVIII inhibitors (BU/mL) for the 2 patients in subgroup 3, patient 15 (A) and patient 16 (B), who developed FVIII-binding IgG1 antibodies and transient low-titer FVIII inhibitors. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). (C-D) Apparent affinity constants of FVIII-binding IgG1 antibodies (mean KA) and FVIII inhibitors (BU/mL) in patient 15 (C) and patient 16 (D). Data for apparent affinity constants include the 95% CIs for up to 2 IgG1 affinity clusters (open blue bars, IgG1 population 1; closed blue bars, IgG1 population 2). The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL).
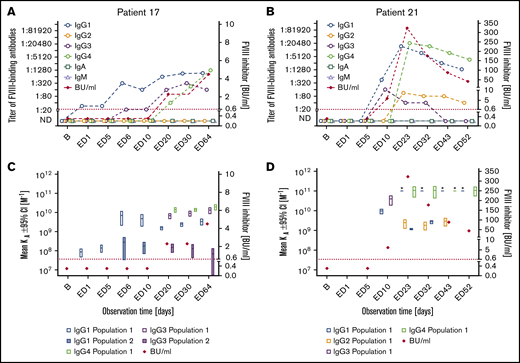
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in 2 representative examples of patients in subgroup 4. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, and IgM, as indicated) and FVIII inhibitors (BU/mL) for 2 representative examples of patients in subgroup 4, patient 17 (A) and patient 21 (B), who developed FVIII-binding IgG subclass-switched antibodies and persistent FVIII inhibitors. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). (C-D) Apparent affinity constants of FVIII-binding antibodies (mean KA), differentiated for individual IgG subclasses and FVIII inhibitors (BU/mL), in patient 17 (C) and patient 21 (D). Data for apparent affinity constants include the 95% CIs for ≤2 affinity clusters for each IgG subclass (open bars, population 1; closed bars, population 2). The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). The asterisk indicates antibodies with apparent affinities that were too high to be assessed. Therefore, they were set to 10e11. The data for the remaining 5 patients of subgroup 4 are shown in supplemental Figure 4.
Longitudinal monitoring of FVIII-binding antibodies and FVIII inhibitors in 2 representative examples of patients in subgroup 4. (A-B) Results of the analysis of FVIII-binding antibodies (IgG1, IgG2, IgG3, IgG4, IgA, and IgM, as indicated) and FVIII inhibitors (BU/mL) for 2 representative examples of patients in subgroup 4, patient 17 (A) and patient 21 (B), who developed FVIII-binding IgG subclass-switched antibodies and persistent FVIII inhibitors. The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). (C-D) Apparent affinity constants of FVIII-binding antibodies (mean KA), differentiated for individual IgG subclasses and FVIII inhibitors (BU/mL), in patient 17 (C) and patient 21 (D). Data for apparent affinity constants include the 95% CIs for ≤2 affinity clusters for each IgG subclass (open bars, population 1; closed bars, population 2). The red dotted lines represent the limit for positive evaluation of FVIII inhibitors (0.6 BU/mL). The asterisk indicates antibodies with apparent affinities that were too high to be assessed. Therefore, they were set to 10e11. The data for the remaining 5 patients of subgroup 4 are shown in supplemental Figure 4.
Comparison of FVIII-binding antibody signatures in patients with persistent FVIII inhibitors and those with nonneutralizing antibodies
In order to compare antibody signatures in patients with FVIII inhibitors (subgroup 4; Table 2) and those with nonneutralizing antibodies (subgroup 2; Table 2), we compared titers and apparent affinity constants of FVIII-binding antibodies, differentiated for IgG subclasses, IgM and IgA (Figure 5). Whereas patients in subgroup 2 (nonneutralizing FVIII-binding antibodies) developed FVIII-binding IgG1 but no other IgG subclass, all patients in subgroup 4 (persistent FVIII inhibitors) developed FVIII-binding IgG1, IgG3, and IgG4 antibodies, and some of them developed FVIII-binding IgG2 antibodies. Moreover, some patients in both subgroups developed FVIII-binding IgA antibodies.
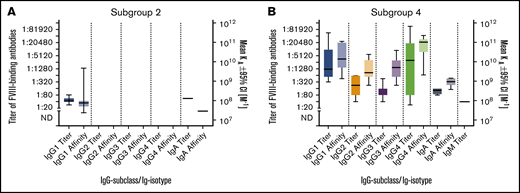
Summary of FVIII-binding antibodies for all patients in subgroups 2 and 4. (A-B) Medians and IQRs for titers and apparent affinity constants of FVIII-binding antibodies, differentiated for IgG subclasses 1-4, IgM and IgA, as detected in patients in subgroup 2, who did not develop FVIII inhibitors (A), and patients in subgroup 4, who developed persistent FVIII inhibitors (B). The calculation of medians and IQRs for titers and apparent affinity constants included all antibody data for each patient in the respective subgroup at each time point analyzed.
Summary of FVIII-binding antibodies for all patients in subgroups 2 and 4. (A-B) Medians and IQRs for titers and apparent affinity constants of FVIII-binding antibodies, differentiated for IgG subclasses 1-4, IgM and IgA, as detected in patients in subgroup 2, who did not develop FVIII inhibitors (A), and patients in subgroup 4, who developed persistent FVIII inhibitors (B). The calculation of medians and IQRs for titers and apparent affinity constants included all antibody data for each patient in the respective subgroup at each time point analyzed.
FVIII-binding IgG1 antibodies differ in their quality between patients with persistent FVIII inhibitors and those with nonneutralizing antibodies
FVIII-binding IgG1 was the only IgG subclass found in both patients with nonneutralizing antibodies (subgroup 2) and patients with persistent FVIII inhibitors (subgroup 4). Therefore, we asked if the quality of FVIII-binding IgG1 antibodies differs between these 2 subgroups. The data indicate that both antibody titers and apparent affinity constants of FVIII-binding IgG1 antibodies were significantly higher in patients with FVIII inhibitors in subgroup 4 (Figure 6).
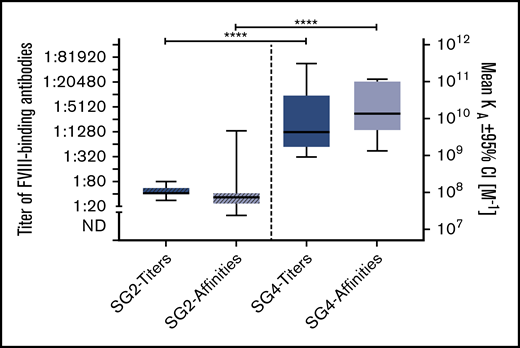
Comparison of titers and apparent affinities for FVIII-binding IgG1 antibodies in patients in subgroups (SG) 2 and 4. Medians and IQRs for titers and apparent affinity constants of FVIII-binding IgG1 antibodies, as detected in patients of subgroup 2, who did not develop FVIII inhibitors, and patients in subgroup 4, who developed persistent FVIII inhibitors. Median IQRs for titers and apparent affinity constants include all antibody data for each patient in the respective subgroup at each time point analyzed. There is a significant difference in both titers and apparent affinity constants between patients of subgroups 2 and 4. ****P < .0001.
Comparison of titers and apparent affinities for FVIII-binding IgG1 antibodies in patients in subgroups (SG) 2 and 4. Medians and IQRs for titers and apparent affinity constants of FVIII-binding IgG1 antibodies, as detected in patients of subgroup 2, who did not develop FVIII inhibitors, and patients in subgroup 4, who developed persistent FVIII inhibitors. Median IQRs for titers and apparent affinity constants include all antibody data for each patient in the respective subgroup at each time point analyzed. There is a significant difference in both titers and apparent affinity constants between patients of subgroups 2 and 4. ****P < .0001.
High-affinity FVIII-binding IgG1 and subsequent development of FVIII-binding IgG3 is a unique antibody signature indicating evolving FVIII inhibitors
Patients who developed FVIII-binding IgG1 and never developed any other FVIII-binding IgG subclass either did not develop FVIII inhibitors or developed transient, low-titer FVIII inhibitors only (Figures 2 and 3; supplemental Figure 3). On the other hand, patients who developed high-affinity FVIII-binding IgG1 antibodies and subsequently FVIII-binding IgG3 antibodies developed persistent FVIII inhibitors that were of high titer in 6 out of 7 patients (Figure 4; supplemental Figure 4). All patients who developed FVIII-binding IgG3 antibodies, subsequently developed high-affinity FVIII-binding IgG4 antibodies. However, FVIII-specific IgG4 antibodies were only detected subsequent to first inhibitor detection (6 out of 7 patients) or at the same time as first FVIII inhibitor detection (1 out of 7 patients) (Figure 4; supplemental Figure 4).
Discussion
HIPS is the first prospective cohort study to evaluate comprehensive changes in the immune system during the first 50 EDs to FVIII in patients with severe hemophilia A. Longitudinal antibody data coming out of this study indicate subpopulations of patients expressing distinct signatures of evolving antibody responses directed against FVIII. These signatures differentiate patients developing persistent FVIII inhibitor responses from those developing nonneutralizing antibody responses and those without any IgG antibody response. Some of the antibody signatures, such as the appearance of FVIII-binding IgG3 antibodies after an initial phase of FVIII-specific IgG1 responses, provide candidates for the development of early biomarkers of FVIII inhibitor development.
Patients in subgroup 1 never developed any FVIII-binding IgG (1-4) antibodies, neutralizing or nonneutralizing. Patients in subgroups 2 and 3 developed FVIII-binding IgG1 antibodies that were either restricted to nonneutralizing antibodies (subgroup 2) or associated with transient FVIII inhibitors (subgroup 3). The underlying immune mechanisms limiting the antibody responses to transient or persistent IgG1 are probably similar in patients of subgroups 2 and 3. It is likely that the epitopes recognized by these IgG1 antibodies determine whether the antibodies are nonneutralizing or neutralizing. Whereas neutralizing antibodies have been shown to be directed against several functional epitopes of FVIII with a predominance of antibodies directed against epitopes of the A2 and C2 domains, nonneutralizing antibodies have been reported to be directed against nonfunctional epitopes of FVIII.24-28 The 7 patients in subgroup 4 developed persistent FVIII inhibitors of mostly high titer, associated with antibody signatures that differed substantially from those observed in patients of subgroups 2 and 3. FVIII-binding IgG1 antibodies appeared first, followed by IgG3 and IgG4 antibodies. The appearance of FVIII-specific IgG3 antibodies after an initial phase of FVIII-specific IgG1 responses was always associated with the subsequent FVIII inhibitor diagnosis and the development of high-affinity FVIII-binding IgG4. In addition to FVIII-binding IgG antibodies, we observed transient low-titer FVIII-binding IgM or IgA in a few patients of subgroups 1, 2, and 4. This finding confirms our previous observations.10,11 The temporary appearance of these antibodies was not associated with any particular FVIII-binding IgG antibody signature or any other clinical parameter.
What differentiates the underlying immune mechanisms that prevented detectable IgG antibody responses against FVIII in patients of subgroup 1, limited the IgG antibody responses to IgG1 in patients of subgroups 2 and 3, and resulted in high-affinity IgG subclass-switched antibodies accompanied by persistent FVIII inhibitor development in patients of subgroup 4? The significantly lower titers and affinity constants of FVIII-binding IgG1 antibodies observed in patients of subgroup 2 compared with patients of subgroup 4 and the lack of FVIII-binding IgG3 and IgG4 responses in patients of subgroups 2 and 3 support the idea that FVIII-directed IgG antibody responses in patients of subgroups 2 and 3 were halted in a less advanced activation phase of FVIII-specific B-cell responses. B-cell activation in response to FVIII could be restrained when second signals, such as those provided by cognate interactions with FVIII-specific helper T cells, are limiting. Recently, Tan et al provided evidence indicating that Nr4a1-3, encoding a small family of orphan nuclear receptors that are rapidly induced by B-cell antigen receptor stimulation, are involved in restraining B-cell responses under conditions of competition for limiting T-cell help.29 The differentiation of B cells into antibody-secreting cells is associated with profound changes in transcriptional programs that are controlled by distinct transcription factors and epigenetic regulators.30 B cells and plasma cells express distinct transcriptomes that are maintained by 2 groups of mutually exclusive transcription factors. One group of transcription factors (eg, Bcl6, PAX-5, and BACH-2) maintains the B-cell program. The other group of transcription factors (eg, Blimp-1, XBP1, and IRF-4) maintains the program of antibody-secreting cells. The mutually exclusive expression programs are maintained by transcriptional repression, such as the B-cell–maintaining transcription factors Bcl6, PAX5, and BACH2, directly suppress the expression of the plasma-cell-maintaining transcription factors, Blimp-1, XBP1, and IRF-4. On the other hand, Blimp-1 represses the expression of Bcl-6 and PAX5.30,31 More recently, molecular mediators that serve as endogenous brakes to the effector B-cell responses, such as antibody class-switch recombination and the development of antibody-secreting cells, were identified in mouse models and in vitro studies.32,33 These brakes control the propensity of B cells to undergo affinity maturation, antibody class-switch recombination, and plasma cell differentiation. Some of these brakes, such as the cytoplasmic aryl hydrocarbon receptor, can act as sensors of immune cells for the local microenvironment.34 A tolerizing local microenvironment could favor the induction of immune tolerance associated with the induction of regulatory T cells and, at the same time, restrict the immune response to a less mature IgG1 response.
The evolving understanding of the molecular regulation of early B-cell effector responses, such as the role of endogenous negative brakes in antibody class-switch recombination and B-cell differentiation into antibody-secreting cells, will help to further unravel the mystery of distinct antibody signatures found in patients with and without FVIII inhibitors following FVIII-replacement therapies. Future studies focusing on the molecular analysis of genome and transcriptome signatures of patients enrolled in the HIPS study will contribute to this process.
For original data, please e-mail the corresponding author, B. M. Reipert (birgit.reipert@fh-krems.ac.at).
Acknowledgments
The authors thank Keith Hoots and Donna DiMichele for their insights in the study design and analysis. They would also thank Fritz Scheiflinger (an employee of Baxalta Innovations at the time of the study), who supported the analytical part of the study, and Elizabeth Donnachie, who did the center qualification of the US clinical centers that participated in the study. The authors are grateful to Evelyn Lockhart Biomedical Communications (BMC) for support with virtual abstract design.
HIPS is a collaborative clinical research study funded by Baxalta Innovations GmbH, a Takeda company, Vienna, Austria.
Authorship
Contribution: B.M.R. designed the immunological program of the study, reviewed and interpreted data, and wrote the manuscript; B.G. managed the immunological analysis, reviewed and interpreted immunological data, and wrote the manuscript; C.J.H. designed the immunological part of the study, established the technology platform for antibody analytics, reviewed and interpreted data, and reviewed the manuscript; V.B. and H.S. ran the antibody analysis, reviewed and interpreted the immunological data, created antibody figures, and reviewed the manuscript; J. Bowen assisted with management and completion of the study, collected clinical data, reviewed data displays, and reviewed and edited manuscript; J. Blatny, K.F., E.S.M., J.K., C. Male, C. McGuinn, V.C.R., M.V.R., M.R., A.D.S., J.M.S., and H.M.Y. cared for study participants and reviewed and edited the manuscript; S.L.M. cared for study participants, reviewed and interpreted data, and reviewed and edited the manuscript; E.S. (coprincipal investigator) cared for study participants, designed the study plan, reviewed and interpreted data and reviewed and edited the manuscript; and D.L.B. (principal investigator) cared for study participants, designed study plan, reviewed and interpreted data, and wrote and reviewed the manuscript.
Conflict-of-interest disclosure: B.M.R. was an employee of Baxalta Innovations at the time of the study but left after study completion. B.G. is an employee of Baxalta Innovations and holds Takeda stock options. C.J.H. was an employee of Baxalta Innovations at the time of the study but is now an employee of Takeda Pharmaceutical (Cambridge, MA) and holds Takeda stock options. V.B. and H.S. received institutional research funding from Baxalta Innovations. J. Bowen received research funding from the study sponsor, University of Texas Health Science Center (Houston, TX). J. Blatny has received consultation/speakers fees from Takeda, Novo Nordisk, Sobi, LFB, Roche, Pfizer, CSL Behring, and Octapharma. K.F. has received unrestricted institutional research grants from CSL Behring and Novo Nordisk and institutional consultancy fees from Grifols, Takeda, Novo Nordisk, and Roche. E.S.M. has received advisory board fees from Bayer and Takeda. C. Male has received institutional fees for study participation from Bayer, Baxalta/Shire/Takeda, Biotest, CSL Behring, Novo Nordisk, and Sobi; an unrestricted institutional grant from Biotest and CSL Behring; and personal honoraria (consultancy, speaker, chair) from Bayer, CSL Behring, Novo Nordisk, and Roche. C. McGuinn received consultancy/advisory board fees from Roche, and Bioverat, Kendrion, Octapharma, BPL. and Biomarin and institutional fees for study participation from Baxalta/Shire/Takeda, Pfizer/Spark, Novo Nordisk, Roche, and Bioverat/Roche, and Bioverativ/Sanofi. S.L.M. received consultancy/advisory board fees from Bayer, Baxalta/Shire/Takeda, Bioverativ/Sanofi, CSL Behring, Roche, and Bioverat, Novo Nordisk, Octapharma, Pfizer, Sangamo, Spark/Roche, and Sobi. V.C.R. received institutional research funding and fees for study participation from Takeda, Pfizer, and Grifols. M.V.R. received research funding and advisory board fees from Alnylam, Biomarin, Bioverativ, Sangamo, and Spark. M.R. received institutional research funding from Bioverativ/Sanofi, Biomarin, Roche, and Bioverat, Novo Nordisk, Shire/Takeda, Spark/Roche, uniQure and consultancy/advisory board fees from Bioverativ/Sanofi, CSL Behring, Roche, and Bioverat, Kedrion, Novo Nordisk, Pfizer, Shire/Takeda, and uniQure. A.D.S. received institutional research funding from Agios, Biomarin, Bioverativ, Daiichi Sankyo, Roche, and Bioverat, Glover Blood Therapeutics, Kendrion, Novartis, Novo Nordisk, OPKO, Octapharma, Pfizer, Prometic, Sangamo, and Takeda and consultant/advisory board fees from Bioverativ, Roche, and Bioverat, Novo Nordisk, Prometic, Sangamo, and Shire/Takeda; no funding was personally accepted (all was paid to the Indiana Hemophilia & Thrombosis Center). J.M.S. received consultancy/advisory boards from Spark, Roche, and Bioverat, Novo Nordisk, Bayer, Takeda, and Sanofi. H.M.Y. received institutional research funding from Novo Nordisk, Takeda, Bayer, Agio, Genentics, and CSL and advisory/consultancy board fees from Novo Nordisk, Takeda, Bayer, Genentics, and Octapharma. E.S. received advisory boards and/or speaker bureaus fees from Bayer, Pfizer, CSL Behring, Novo Nordisk, Shire/Takeda, Sobi, Bioverativ, Grifols, Kedrion, Octapharma, Roche, Spark, and uniQure. D.L.B. received research funding from Baxalta/Takeda. J.K. declares no competing financial interests.
Correspondence: B. M. Reipert, IMC University of Applied Sciences Krems, Piaristengasse 1, 3500 Krems, Austria; e-mail: birgit.reipert@fh-krems.ac.at.

