

Key Points
The antioxidant NAC extends survival of the Vav-Cre:JAK2V617F knockin murine model of polycythemia vera.
NAC has the potential to inhibit thrombosis caused by the JAK2V617F mutation.

Abstract
Thrombosis is a major cause of mortality in patients with myeloproliferative neoplasms (MPNs), though there is currently little to offer patients with MPN beyond aspirin and cytoreductive therapies such as hydroxyurea for primary prevention. Thrombogenesis in MPN involves multiple cellular mechanisms, including platelet activation and neutrophil-extracellular trap formation; therefore, an antithrombotic agent that targets one or more of these processes would be of therapeutic benefit in MPN. Here, we treated the JAK2V617F knockin mouse model of polycythemia vera with N-acetylcysteine (NAC), a sulfhydryl-containing compound with broad effects on glutathione replenishment, free radical scavenging, and reducing disulfide bonds, to investigate its antithrombotic effects in the context of MPN. Strikingly, NAC treatment extended the lifespan of JAK2V617F mice without impacting blood counts or splenomegaly. Using an acute pulmonary thrombosis model in vivo, we found that NAC reduced thrombus formation to a similar extent as the irreversible platelet inhibitor aspirin. In vitro analysis of platelet activation revealed that NAC reduced thrombin-induced platelet-leukocyte aggregate formation in JAK2V617F mice. Furthermore, NAC reduced neutrophil extracellular trap formation in primary human neutrophils from patients with MPN as well as healthy controls. These results provide evidence that N-acetylcysteine inhibits thrombosis in JAK2V617F mice and provide a pre-clinical rationale for investigating NAC as a therapeutic to reduce thrombotic risk in MPN.
Introduction
Myeloproliferative neoplasms (MPNs) are a group of chronic hematologic malignancies that include polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis and are characterized by the overproduction of mature myeloid cells. The constitutive activating mutation in Janus kinase 2 (ie, JAK2V617F) is the most common mutation found in MPN patients. Individuals harboring JAK2V617F mutations are at high risk for thrombosis, which is a major cause of morbidity and mortality in patients with MPN. Thus, the primary goal of treatment in PV and ET is to reduce thrombotic risk. The current arsenal of agents to reduce thrombotic risk in MPN includes aspirin and cytoreductive agents such as hydroxyurea. Aspirin is an irreversible platelet inhibitor, and current National Comprehensive Cancer Center Network guidelines recommend low-dose aspirin (81-100 mg daily) for all JAK2-mutated patients with MPN who are not at high risk of bleeding due to acquired von Willebrand disease.1 Although aspirin has been established to reduce the risk of thrombotic events in patients with PV,2 in low-risk patients with ET (JAK2 negative and without cardiovascular risk factors), antiplatelet therapy with aspirin does not reduce the incidence of thrombotic events.3 Cytoreductive agents such as hydroxyurea or anagrelide are given to high-risk patients (age >60 years or previous thrombosis) to reduce thrombotic risk. However, in patients with ET aged 40 to 59 years and lacking high-risk factors for thrombosis or extreme thrombocytosis, preemptive addition of hydroxyurea to aspirin does not reduce vascular events.4 Moreover, cytoreductive drugs are not without significant side effects, including gastrointestinal toxicities, edema, palpitations, skin ulcers, increased risk of skin cancer, and potentially promotion of leukemia. Thus, treatments that reduce thrombotic risk in MPN without significant side effects would improve the quality of life and outcome in patients with MPN, particularly in younger patients with MPN.
Since the discovery of the JAK2V617F mutation in 2005,5-9 numerous mouse models have been developed to study MPN in vivo.10 In the conditional Vav-Cre:JAK2V617F knockin mouse model of MPN, mice die at ∼3 months of age, presumably from thrombosis, given that death is sudden and without evident preceding decline in health.11 These mice have elevated blood counts and splenomegaly but do not develop bone marrow fibrosis. Thus, this JAK2V617F knockin mouse model is ideal to identify therapeutics that can be used to prevent thrombosis in MPN.
Multiple cell types, including platelets, leukocytes, and endothelial cells, participate in the formation of a blood clot. Damaged endothelium exposes collagen to platelets, leading to their activation and formation of a plug. There is a growing appreciation for the crucial role of leukocytes in thrombus initiation and progression, as platelet activation can cause activation of leukocytes (most commonly of neutrophils), leading to formation of platelet-leukocyte aggregates (PLAs). A growing body of evidence supports the notion that aberrant leukocyte activation plays an important role in thrombosis in MPN.12 Interestingly, an elevated leukocyte count, but not platelet count,13 correlates with thrombotic risk and cardiovascular disease14 in MPN. Moreover, elevated levels of circulating PLAs are correlated with thrombotic risk in MPN.15
Formation of PLAs can induce neutrophil extracellular trap (NET) formation (NETosis), a phenomenon dependent on oxidative stress whereby neutrophils release chromatin lined with granular components with antimicrobial properties. The primary function of NETs is to entrap pathogens for host defense, but they can also bind platelets and red blood cells, contributing to thrombosis. Neutrophils from MPN mouse models as well as patients with MPN are primed for NETosis,12 implicating involvement of NETs in the prothrombotic phenotype of MPN. Thus, preventing leukocyte activation and NETosis in MPN may be an effective approach to reduce thrombosis. Agents that not only act as reactive oxygen species (ROS) scavengers but also have antithrombotic properties with a favorable safety profile/minimal side effects would be attractive therapeutics for the prevention of thrombosis in MPN.
N-acetylcysteine (NAC) is an agent that has antioxidant, mucolytic, and anti-inflammatory properties. It is currently used for acetaminophen overdose and contrast nephropathy prophylaxis and as a mucolytic agent in cystic fibrosis. It has proven safe even at the large doses of >10 g given for acetaminophen overdose. NAC is being investigated as a therapy for thrombotic diseases such as thrombotic thrombocytopenic purpura (NCT01808521). Given that NETosis is dependent on oxidative stress, NAC has been found to block NETosis in vitro.16 Therefore, NAC may prevent NETosis in MPN, leading to reduced thrombosis. NAC has also been shown to be thrombolytic in arterial thrombi17 and to reduce the formation of PLAs in a model of experimental diabetes.18 In addition, NAC has anti-inflammatory properties and decreases serum inflammatory cytokines,19 which would also be beneficial in MPN. To that end, we tested the efficacy of oral treatment with NAC in JAK2V617F knockin mice with respect to their lifespan, thrombosis, and parameters such as blood counts, splenomegaly, and tissue pathologies. We found that NAC reduces thrombosis in these mice via inhibition of PLA and NET formation. Therefore, NAC may represent a low-risk therapy for the reduction of thrombosis in MPN and should be investigated in human studies.
Materials and methods
Mice
All animal procedures were performed under approval of the Institutional Animal Care and Use Committee at the University of California, Irvine. Equal numbers of male and female mice were used for all experiments. We used the Vav-Cre:JAK2V617F murine model that has been previously described.11 Mice were given drinking water with added NAC (2 g/L) or aspirin (16 mg/L) beginning at 4 to 6 weeks of age, and this was replenished weekly. This dose of aspirin (16 mg/L) translates to 3.2 mg/kg per day, assuming the average mouse weighs 20 g and drinks 4 mL of water per day. Since aspirin is used in a wide range of doses ranging from 0.15 to 60 mg/kg in mice, we chose a low-dose aspirin treatment, since patients with MPN are prone to both thrombotic and hemorrhagic events. Complete blood counts were measured using the ABCVet Hemalyzer (scil) using blood diluted 1:1 with 100 mM EDTA. Spleen and liver weights were recorded upon euthanasia or death.
Acute pulmonary thrombosis model
An in vivo acute pulmonary thrombosis model was performed as previously described.20 Briefly, mice were anesthetized with isoflurane and then injected IV with a mixture of collagen (0.8 mg/kg) and epinephrine (6 mg/kg). Circulating platelet counts were recorded using the ABCVet Hemalyzer 1 hour before and 5 minutes after treatment with collagen/epinephrine. Survival of mice was recorded 30 minutes after treatment. Immediately following death (or 30 minutes after treatment), lungs were perfused with phosphate-buffered saline (PBS) and fixed in 10% zinc formalin for histological processing. At least 3 nonoverlapping images per lung section were taken using the BZ-X700 Keyence bright-field microscope. Thrombi were measured and quantified by an investigator blinded to the conditions.
PLAs
All experiments involving human blood were performed under institutional review board approval from the University of California, Irvine. We used whole-blood flow cytometry to detect PLAs using fluorochrome-conjugated monoclonal antibodies to identify platelets and leukocytes. Briefly, 5 µL whole blood was diluted in 55 µL modified N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer without calcium (145 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.5 mM NaHPO4, 5 mM glucose, and 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid sodium) within 30 minutes of collection. Diluted blood was gently mixed with an antibody mix containing 0.2 µg CD41-APC (MWReg30;Thermo Fisher Scientific) and 0.2 µg CD45-PerCP-Cy5.5 (30-F11; BioLegend) and incubated for 20 minutes at room temperature, protected from light. To measure PLA subtypes, blood was also incubated with Alexa Fluor 488-Ly6G (1A8; BioLegend) and PE-CD11b (M1/70; BioLegend). For agonist-induced and inhibition experiments, thrombin at 1 U/mL (MilliporeSigma) was used. 1xRBC lyse/fix solution (BioLegend) was then added and further incubated for 15 minutes. Acquisition was performed within 30 minutes after RBC lysis. The percentage of CD45+/CD41+ double-positive events representing PLAs was recorded.
In vitro NET formation
To assess the effect of NAC on NET formation in vitro, neutrophils were isolated using the EasySep Direct Human Neutrophil Isolation Kit according to the vendor’s protocol (StemCell Tech, Vancouver, BC, Canada). Neutrophils were resuspended in RPMI 1640 containing 2% fetal bovine serum (Thermo Fisher Scientific) then were seeded at 2 × 105 cells/chamber on Permanox plastic chamber slides coated with Poly-L-Lysine (Thermo Fisher Scientific). Neutrophils were incubated at 37°C with 1 mM NAC for 30 minutes before stimulation with 10 nM phorbol 12-myristate 13-acetate (PMA) for 3 to 6 hours at 37°C. Cells were fixed with 2% paraformaldehyde and incubated overnight at 4°C. Immunofluorescence was performed to detect the NET marker citrullinated histone 3 (H3cit) as previously described.12 Briefly, cells were washed 3 times with PBS, permeabilized with 0.1% Triton X-100 + 0.1% sodium citrate for 10 minutes at 37°C, and then blocked with 3% bovine serum albumin for 90 min at 37°C. Cells were incubated with 1:1000 rabbit anti-H3cit overnight at 4°C and then with 1:2000 Alexa 488 donkey anti-rabbit immunoglobulin G for 2 hours at 25°C (ab5103 and ab150073, respectively; Abcam). DNA was counterstained with DAPI, and then slides were coverslipped with Permafluor mountant (Thermo Fisher Scientific). Micrographs were taken with the BZ-X700 Keyence fluorescence microscope. NETs were quantified based on H3cit positivity or by swollen/delobular nuclei or extracellular DNA web-like strands. Percentages of NETs were determined based on 3 to 5 images per condition or >50 neutrophils per condition. NETs were quantified by 2 investigators blinded to the conditions.
SYTOX-based detection of extracellular DNA
We seeded 1 × 105 purified neutrophils/well into a cell imaging 96-well plate (Eppendorf) and then added 1 µM SYTOX green nucleic acid stain (Thermo Fisher Scientific). Cells were pretreated and stimulated similar to immunofluorescence assays. Plates were incubated at 37°C, 5% CO2 in a Biotek Cytation 5 imaging plate reader, and fluorescence was measured every 15 min at 504/523 nm for up to 16 hours. NETs were measured by fluorescence as a result of extracellular DNA release.
Statistical analysis
All statistical analyses were performed using GraphPad Prism v7.0 (GraphPad, La Jolla, CA). For survival analyses as represented by Kaplan-Meier curves, a log-rank (Mantel-Cox) test was performed. Other statistical analyses were performed using a 1-way analysis of variance (ANOVA) (Sidak’s multiple comparison test) or 2-way ANOVA (Tukey’s test). Significant differences were indicated as *P < .05, **P < .01, ***P < .001, and ****P < .0001. All experiments were performed ≥3 independent times unless otherwise stated.
Results
NAC extends the lifespan of JAK2V617F knockin mice
To investigate the effect of NAC on MPN pathology, in particular thrombosis, we added NAC (2 g/L) or aspirin (16 mg/L) to the drinking water of JAK2V617F knockin mice at 2 months of age and maintained them on this water continuously thereafter. Consistent with previous reports, JAK2V617F knockin mice begin to die at ∼3 months of age without signs of preceding illness, consistent with thrombosis as the cause of death.11 In support of this, we observed vascular occlusions in the lungs of JAK2V617F, but not wild-type (WT) mice (supplemental Figure 1). Strikingly, treatment with NAC extended the lifespan of JAK2V617F mice past 12 months of age (Figure 1A). Aspirin, however, did not increase the lifespan of these mice (Figure 1A). NAC had no effect on spleen or liver weights (Figure 1B-C) or spleen, liver, or bone marrow architecture (supplemental Figure 1). In addition, NAC had no effect on blood counts in WT or JAK2V617F knockin mice (Figure 1D-F). These data suggest that NAC extends the lifespan of mice with MPN through a mechanism independent of cytoreduction. Based on these data, we postured that NAC may be inhibiting thrombus formation in JAK2V617F knockin mice.
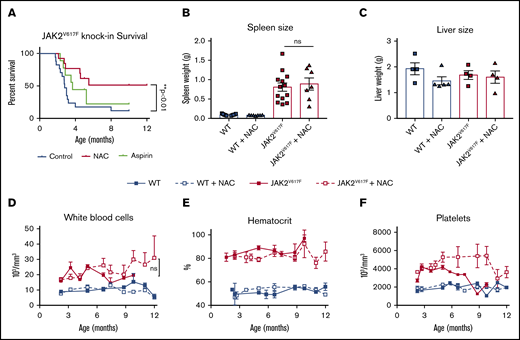
NAC extends the lifespan in a JAK2V617Fknockin mouse model of MPN. (A) Kaplan-Meyer survival curve representing JAK2V617F mice whose drinking water contained NAC (2 g/L) or aspirin (16 mg/L), **P < .01 log-rank test. Spleen (B) and liver (C) weights at euthanasia or death. (D-F) Peripheral blood counts, including white blood cell count (D), hematocrit (E), and platelet count (F), in WT or JAK2V617F mice treated with plain water or NAC-containing water. Error bars represent standard error of the mean (SEM); n = 6-12 per group. Shown are representative data from 2 biological replicates. ns, not significant.
NAC extends the lifespan in a JAK2V617Fknockin mouse model of MPN. (A) Kaplan-Meyer survival curve representing JAK2V617F mice whose drinking water contained NAC (2 g/L) or aspirin (16 mg/L), **P < .01 log-rank test. Spleen (B) and liver (C) weights at euthanasia or death. (D-F) Peripheral blood counts, including white blood cell count (D), hematocrit (E), and platelet count (F), in WT or JAK2V617F mice treated with plain water or NAC-containing water. Error bars represent standard error of the mean (SEM); n = 6-12 per group. Shown are representative data from 2 biological replicates. ns, not significant.
NAC inhibits thrombus formation in vivo
To investigate the effect of NAC on thrombosis, we used an acute pulmonary thrombosis model as previously described.20 This model involves activating platelets in vivo using a mixture of collagen and epinephrine then recording survival and histologically analyzing lung tissues for thrombi. Circulating platelet counts were measured immediately before and 5 minutes following collagen/epinephrine injection (Figure 2A). As expected, the number of circulating platelets was significantly reduced in mice treated with collagen/epinephrine compared with untreated controls (P < .001, 2-way ANOVA), suggesting a thrombus formation in the lungs (Figure 2A). Aspirin irreversibly inhibits platelet activation and was used as a positive control in this assay. As expected, aspirin-treated mice had significantly higher circulating platelets compared with PBS-treated mice following collagen/epinephrine treatment (P < .01, 2-way ANOVA; Figure 2A). Strikingly, NAC-treated mice phenocopied aspirin-treated mice with a significantly higher circulating platelet count following collagen/epinephrine treatment compared with PBS controls (P < .01, 2-way ANOVA; Figure 2A). All mice (6 of 6) treated with collagen/epinephrine alone died within 30 minutes after injection as a result of the pulmonary thrombi formation (Figure 2B). However, 50% of NAC-treated mice (3 of 6) survived collagen/epinephrine treatment while 33% of aspirin-treated mice (2 of 6) survived collagen/epinephrine treatment (Figure 2B). Upon histological analysis of the lungs, pulmonary thrombi were clearly visible in mice treated with the collagen/epinephrine mixture, with an average of 4 thrombi per field (Figure 2C,E). There were significantly fewer visible thrombi in NAC-treated mice (1.5 thrombi/field; P < .01, 2-way ANOVA) as well as aspirin-treated mice (1.6 thrombi/field; P < .01, 2-way ANOVA) compared with collagen/epinephrine treatment alone (Figure 2C,F-G). These data indicate that NAC inhibits thrombus formation induced by collagen and epinephrine-induced platelet activation as well as aspirin in vivo.
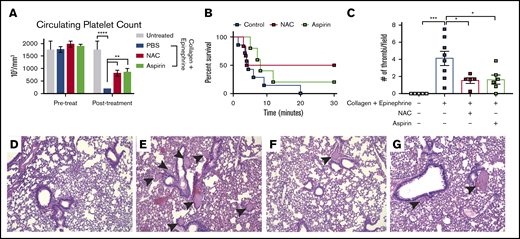
NAC reduces pulmonary thrombus formation in vivo. (A) Platelet counts in adult mice 1 hour before and 5 minutes following IV injection with a mixture of collagen (0.8 mg/kg) + epinephrine (6 mg/kg). (B) Kaplan-Meyer curve representing survival of mice injected with collagen + epinephrine mixture to induce a thromboembolism. Survival was recorded as 30 minutes after injection. (C) Quantification of pulmonary thrombi per image field. (D-G) Representative histology images of lungs from untreated control (D), collagen/epinephrine-treated (E), NAC + collagen/epinephrine (F), or aspirin + collagen/epinephrine (G). Pulmonary thrombi are indicated by black arrowheads; n = 6 mice per group, original magnification ×20. *P < .05, **P < .01, ***P < .001, and ****P < .0001, 2-way ANOVA. Error bars represent SEM. Shown are representative data from 3 biological replicates.
NAC reduces pulmonary thrombus formation in vivo. (A) Platelet counts in adult mice 1 hour before and 5 minutes following IV injection with a mixture of collagen (0.8 mg/kg) + epinephrine (6 mg/kg). (B) Kaplan-Meyer curve representing survival of mice injected with collagen + epinephrine mixture to induce a thromboembolism. Survival was recorded as 30 minutes after injection. (C) Quantification of pulmonary thrombi per image field. (D-G) Representative histology images of lungs from untreated control (D), collagen/epinephrine-treated (E), NAC + collagen/epinephrine (F), or aspirin + collagen/epinephrine (G). Pulmonary thrombi are indicated by black arrowheads; n = 6 mice per group, original magnification ×20. *P < .05, **P < .01, ***P < .001, and ****P < .0001, 2-way ANOVA. Error bars represent SEM. Shown are representative data from 3 biological replicates.
NAC rescues mice containing JAK2V617F cells from an induced thrombosis
Given that Vav-Cre expression can be found in endothelial cells, we used a transplantation approach to interrogate how JAK2V617F expression in hematopoietic cells alone influences thrombogenesis and the impact of NAC on this. We transplanted equal numbers of JAK2V617F and WT bone marrow cells into lethally irradiated WT recipients and performed acute pulmonary thrombosis assays 3 months after transplant. Compared with mice transplanted with WT cells alone (WT:WT), mice containing JAK2V617F cells (VF:WT) were more susceptible to induced pulmonary thrombosis in response to thrombotic agents (Figure 3). All VF:KI mice (6 of 6) treated with 1× dose or 1/2× dose of collagen/epinephrine alone died within 30 minutes, while 25% (1 of 4) of WT:WT mice survived (Figure 3A-B). At 1/4× dose of collagen/epinephrine, 83% (5 of 6) WT:WT mice survived, while only 33% (1 of 3) of VF:WT mice survived (Figure 3C). These data suggest that mice containing JAK2V617F hematopoietic cells are hypersensitive to an induced thrombosis. Similar to WT mice, we observed that NAC significantly prolonged survival of VF:WT mice following induction of thrombosis with collagen/epinephrine (Figure 3A). While 100% of VF:WT mice ultimately succumbed to death from collagen/epinephrine at full dose, NAC prolonged survival from 3.75 minutes after induction in controls to over 14 minutes after induction with NAC (P = .01, log-rank test; Figure 3A). NAC also increased the percentage of VF:WT mice surviving to 50% with the 1/2× dose of collagen/epinephrine (Figure 3B). Concurrent with the survival trend, we observed fewer thrombi in lung tissues of NAC and aspirin-treated VF:WT mice compared with controls (Figure 3D-G).
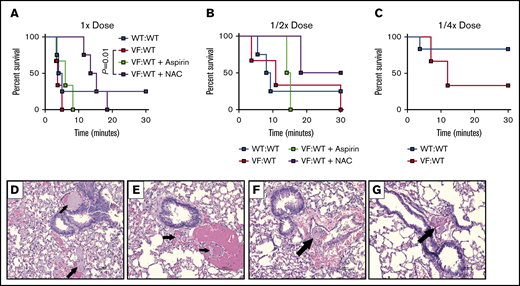
NAC improves survival following induction of acute thrombosis in mice transplanted with JAK2V617Fcells. Control (WT:WT) and JAK2V617F engrafted (VF:WT) mice were pretreated with aspirin (50 mg/kg) or NAC (400 mg/kg) for 1 hour, and then treated with decreasing concentrations of a mixture of collagen and epinephrine via retro-orbital injection. (A-C) Kaplan-Meyer curve representing survival of mice injected with collagen and epinephrine concentrations of 0.8 mg/kg and 6 mg/kg (A), 0.4 mg/kg and 3 mg/kg (B), and 0.2 mg/kg and 1.5 mg/kg (C), respectively. (D-G) Representative hematoxylin and eosin–stained lung tissues from collagen and epinephrine-injected mice. Treatments include control (WT:WT) (A), VF:WT (B), VF:WT + NAC (C), and VF:WT + aspirin (D). Arrows represent thrombi. P = .01, log-rank test. n = 3-4 mice per group, original magnification ×20. Shown are representative data from 2 biological replicates.
NAC improves survival following induction of acute thrombosis in mice transplanted with JAK2V617Fcells. Control (WT:WT) and JAK2V617F engrafted (VF:WT) mice were pretreated with aspirin (50 mg/kg) or NAC (400 mg/kg) for 1 hour, and then treated with decreasing concentrations of a mixture of collagen and epinephrine via retro-orbital injection. (A-C) Kaplan-Meyer curve representing survival of mice injected with collagen and epinephrine concentrations of 0.8 mg/kg and 6 mg/kg (A), 0.4 mg/kg and 3 mg/kg (B), and 0.2 mg/kg and 1.5 mg/kg (C), respectively. (D-G) Representative hematoxylin and eosin–stained lung tissues from collagen and epinephrine-injected mice. Treatments include control (WT:WT) (A), VF:WT (B), VF:WT + NAC (C), and VF:WT + aspirin (D). Arrows represent thrombi. P = .01, log-rank test. n = 3-4 mice per group, original magnification ×20. Shown are representative data from 2 biological replicates.
As platelet activation is a key component of thrombosis, we first investigated whether NAC may prevent thrombosis by inhibiting platelet activation. Platelets from JAK2V617F mice did not show basal hyperactivation as measured by P-selectin exposure, αIIbβ3 integrin activation or phosphatidylserine exposure and also did not show increased agonist-induced activation as compared with platelets from WT mice (supplemental Figure 2). NAC treatment had no marked effect on agonist-induced integrin activation and P-selectin (CD62P) exposure on WT or JAK2V617F platelets (supplemental Figure 2). NAC had no effect on ionomycin-stimulated phosphatidylserine exposure in WT or mutant platelets (supplemental Figure 3). These observations suggest that spontaneous and fatal thrombus formation in JAK2V617F knockin mice is not a result of a hyperactive platelet phenotype and that NAC has no marked effect on platelet activation responses.
NAC inhibits PLA formation in vitro
High levels of circulating PLAs are linked to thrombosis and inflammation. To investigate how NAC impacts PLA formation in vitro, whole blood obtained from mice on NAC or plain water was stimulated with thrombin and then PLAs and platelet-neutrophil aggregates (PNAs) were measured by flow cytometry (Figure 4A). PLAs were also observed in murine blood smears following stimulation with thrombin (indicated by the red arrowhead in Figure 4B-C). We observed a significant increase in PLAs and PNAs following thrombin stimulation in both WT and JAK2V617F knockin mice on plain water (Figure 4D-E). In WT mice on NAC water, thrombin stimulation resulted in a significant increase in PLA and PNA formation (P < .05; Figure 4D-E). However, in JAK2V617F mice on NAC water, there was no significant increase in PLA and PNA formation following thrombin stimulation (Figure 4D-E), suggesting that NAC inhibits PLA formation in JAK2V617F knockin mice. Given that JAK2V617F knockin mice most closely resemble a PV phenotype, we measured circulating PLA and PNA levels in PV patients and normal controls. We observed a significant increase in circulating PLAs in patients with PV (mean 9.0% ± 1.1%, n = 17) compared with normal controls (mean 5.2% ± 0.7%, n = 13; Figure 4F, P = .00095, Student t test). Similarly, we observed an increase in circulating PNAs in patients with PV compared with normal controls, though this trend did not reach significance (Figure 4F-G, P = .17, Student t test). Taken together, these data suggest that NAC may reduce thrombosis and extend lifespan in JAK2V617F knockin mice in part through its ability to block platelet activation-induced PLA formation.
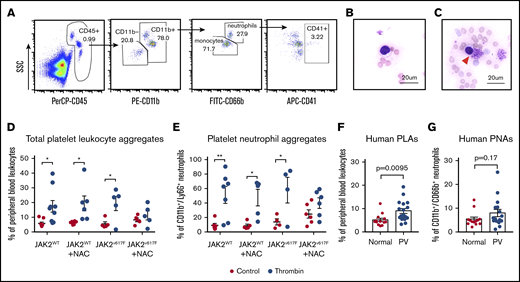
NAC inhibits thrombin-induced PLA formation in JAK2V617Fknockin mice. (A) Representative gating strategy to measure circulating or thrombin-stimulated PLAs (based on CD45+/CD41+ double-positive events). PNAs are defined as CD45+/CD11b+/CD66b+ (for human) or Ly6G+ (for mice)/CD41+ events. (B) Bright-field image of unstimulated murine blood smear stained with Wright Giemsa. (C) Bright-field image of PLA (denoted by red arrowhead) from murine blood stimulated with 2 U thrombin. Original magnification ×60; scale bars represent 20 μm. (D-E) Quantification of PLAs (D) or PNAs (E) in WT or JAK2V617F knockin mice with or without NAC treatment. *P < .05, **P < .01, 2-way ANOVA. (F-G) Circulating PLAs (F) or PNAs (G) in fresh human blood from patients with PV and healthy donors. Each data point represents an individual. Error bars represent SEM. Shown are representative data from 3 biological replicates. SSC, side scatter.
NAC inhibits thrombin-induced PLA formation in JAK2V617Fknockin mice. (A) Representative gating strategy to measure circulating or thrombin-stimulated PLAs (based on CD45+/CD41+ double-positive events). PNAs are defined as CD45+/CD11b+/CD66b+ (for human) or Ly6G+ (for mice)/CD41+ events. (B) Bright-field image of unstimulated murine blood smear stained with Wright Giemsa. (C) Bright-field image of PLA (denoted by red arrowhead) from murine blood stimulated with 2 U thrombin. Original magnification ×60; scale bars represent 20 μm. (D-E) Quantification of PLAs (D) or PNAs (E) in WT or JAK2V617F knockin mice with or without NAC treatment. *P < .05, **P < .01, 2-way ANOVA. (F-G) Circulating PLAs (F) or PNAs (G) in fresh human blood from patients with PV and healthy donors. Each data point represents an individual. Error bars represent SEM. Shown are representative data from 3 biological replicates. SSC, side scatter.
NAC inhibits NETs in vitro
Activation of neutrophils results in NETosis and this phenomenon has been shown to promote thrombosis in MPN.12 To investigate the role of NAC on NET formation, we first assessed NETosis in vitro in human neutrophils by immunofluorescence (Figure 5). We confirmed that PMA is a strong activator of neutrophils and rapidly induces NET formation in neutrophils from MPN patients and normal controls, as measured by H3cit positivity and DNA morphology (P < .01 and P < .0001, respectively, 1-way ANOVA; Figure 5A). NAC reduced PMA-induced NET formation in both normal and MPN neutrophils (Figure 5B-C). We also used a fluorescence-based assay to measure extracellular DNA stained with the nucleic acid dye SYTOX as a measure for NETosis (Figure 5D-E). A representative time course of PMA-induced NETosis from stimulated healthy neutrophils is shown in Figure 5D. Quantification of SYTOX relative fluorescence units (RFUs) were normalized to unstimulated healthy neutrophil fluorescence (Figure 5E). We found a significant increase in RFU following PMA stimulation in both normal and MPN neutrophils (P < .00001 and P < .001, respectively, Student t test; Figure 5E). Pretreatment with NAC resulted in a significant decrease in PMA-induced NETs in MPN neutrophils (Figure 5E). To assess the impact of platelets on NET formation, we coincubated neutrophils with platelets that were activated with thrombin (supplemental Figure 5). Consistent with other reports that showed activated platelets induce NET formation,21,22 we observed a modest increase in NET formation in normal as well as MPN neutrophils upon coincubation with activated normal platelets (supplemental Figure 5). MPN platelets, however, induced NET formation even without ex vivo activation with thrombin (supplemental Figure 5). These data suggest that MPN platelets may be more apt to bind to and activate neutrophils, leading to NET formation, which contributes to the prothrombotic state in MPN.
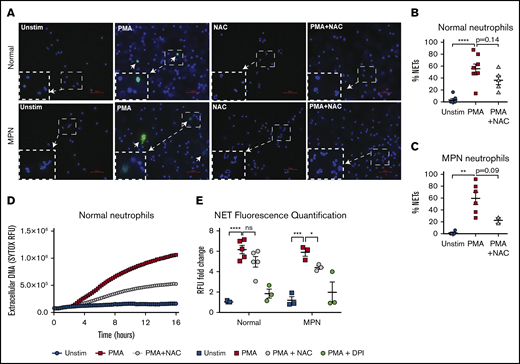
NETs are reduced by NAC in MPN patients. (A) Representative immunofluorescence images of NETs from normal and MPN individuals. H3cit is shown in green, and DAPI-stained DNA is shown in blue. Arrows represent H3cit+ NETs. Bars represent 50 μm. Original magnification ×40. (B-C) Quantification of NETs from normal individuals (B) or patients with MPN (C) upon stimulation with 10 nM PMA and PMA + NAC; **P < .01, ****P < .0001 1-way ANOVA. (D) Representative time course of extracellular DNA via RFUs as measured by the nucleic acid dye SYTOX. (E) Quantification of RFUs normalized to unstimulated normal neutrophils. The inhibitor diphenyleneiodonium (DPI) was used as a positive control for NET inhibition. Each point represents an individual; *P < .05, ***P < .001, ****P < .0001, Student t test. Error bars represent SEM. Shown are representative data from 3 biological replicates.
NETs are reduced by NAC in MPN patients. (A) Representative immunofluorescence images of NETs from normal and MPN individuals. H3cit is shown in green, and DAPI-stained DNA is shown in blue. Arrows represent H3cit+ NETs. Bars represent 50 μm. Original magnification ×40. (B-C) Quantification of NETs from normal individuals (B) or patients with MPN (C) upon stimulation with 10 nM PMA and PMA + NAC; **P < .01, ****P < .0001 1-way ANOVA. (D) Representative time course of extracellular DNA via RFUs as measured by the nucleic acid dye SYTOX. (E) Quantification of RFUs normalized to unstimulated normal neutrophils. The inhibitor diphenyleneiodonium (DPI) was used as a positive control for NET inhibition. Each point represents an individual; *P < .05, ***P < .001, ****P < .0001, Student t test. Error bars represent SEM. Shown are representative data from 3 biological replicates.
Discussion
Here, we demonstrate that NAC extends lifespan in the Vav-Cre–inducible JAK2V617F knockin mouse model of PV. Consistent with previous reports, these mice have erythrocytosis, leukocytosis, and splenomegaly (Figure 1; supplemental Figure 1).11,12 We observed thrombocytosis in our mice, as would be expected for a PV phenotype, although an elevated platelet count was not observed in the original description of this model by Mullally et al.11 Mice die suddenly without preceding illness and are found to have thrombi in the lungs at necropsy, implicating thrombosis as the cause of death in this model. However, we cannot overlook the possibility that these mice may also die of other causes, for example from hemorrhagic events, which may explain why NAC did not rescue all JAK2V617F mice. In addition, the Vav-Cre–inducible model does have expression of JAK2V617F in endothelial cells, this study does not address the impact of NAC specifically on endothelial cells. Nonetheless, our findings corroborate past work using the JAK2V617F knockin mouse model for antithrombotic studies and demonstrate for the first time an intervention that can extend survival of JAK2V617F knockin mice.
We found that NAC did not reduce blood counts or splenomegaly in JAK2V617F knockin mice; however, Marty et al23 previously reported that NAC reduced spleen weight and blood counts and reduced the frequency of JAK2V617F progenitors in a competitive transplantation JAK2V617F murine model. The impact of NAC on blood counts and spleen size by Marty et al23 could possibly be explained by the selective reduction of JAK2V617F cells. Because hematopoiesis in our model is made up entirely of JAK2V617F cells, this could explain why we did not see the same impact on blood counts or spleen weight as Marty et al.23
Using an acute pulmonary thrombosis model in WT mice as well as mice transplanted with JAK2V617F hematopoietic cells, we found that NAC reduces thrombus formation in vivo. While NAC was as effective as aspirin at reducing an induced thrombus in WT mice, aspirin did not improve survival of JAK2V617F knockin mice, suggesting that NAC has additional benefits in MPN that are distinct from aspirin. Our data suggest that classical platelet activation is not the primary mechanism of thrombogenesis in this PV mouse model and are in line with the lack of correlation between platelet count and thrombotic risk in patients with MPN.3 In fact, although platelet number and size were significantly elevated in the JAK2V617F knockin mice, this did not translate to a prothrombotic phenotype, as platelet activation in response to agonists was unaltered by JAK2V617F expression. Interestingly, the model with Mx1-Cre–inducible human transgene of JAK2V617F knockin with an ET phenotype showed heightened platelet responses in vitro and decreased tail bleeding time.24 One possible explanation could be due to the different mouse models used. Moreover, another model of JAK2V617F knockin showed blunted platelet responses in vitro but rapid and unstable thrombus formation in vivo.25 Similarly, platelet-specific (Pf4-Cre) expression of JAK2V617F did not result in a prothrombotic phenotype, while Tie2-Cre inducible JAK2V617F caused dysfunctional hemostasis and carotid artery thrombosis.26 Other groups using the same Vav-Cre–inducible JAK2V617F mouse model showed significantly increased predisposition for venous thrombosis in engrafted mice despite defective platelet responses in vitro.12 We too observed increased mortality of JAK2V617F engrafted mice in the pulmonary thrombosis model, although platelet reactivity was unchanged.
We found that platelets from JAK2V617F mice do not exhibit aberrant activation in vitro, as assessed by conventional assays such as P-selectin, integrin, or phosphatidylserine exposure at baseline or with stimulation. However, we found that platelets from patients with MPN modestly induce NETs from both normal and MPN neutrophils without ex vivo stimulation, whereas normal control platelets only activate NETs with the addition of ex vivo stimulation. Our findings suggest that MPN platelets may play a crucial role in promoting NETosis in MPN, highlighting the complex interplay of these cells to induce NETosis.
In this study, we observed no marked difference in PMA-induced NETosis using purified neutrophils from patients with MPN vs healthy controls. However, Wolach et al showed that MPN neutrophils exhibited elevated ionomycin-induced NETosis.12 Our results highlight the importance of stimulant type to induce differential NETosis between MPN and unaffected individuals. Certain types of NETosis, such as PMA-induced NETosis, are dependent upon ROS and are inhibited by NAC.27 While oxidative stress has been linked to NETs, we did not observe elevated levels of ROS at baseline or following stimulation in MPN patient leukocytes or in JAK2V617F knockin mice compared with healthy controls (supplemental Figure 4). Lower ROS levels that we have observed in leukocytes in patients with MPN could be attributed to an increased antioxidant potential in JAK2V617F CD34+ progenitors.28
Our study does not specifically address the impact of NAC on established thrombosis. Interestingly, NAC has been investigated for its thrombolytic effects in experimental models of acute ischemic stroke and was shown to promote lysis of arterial thrombi via reduction of disulfide bridges in large von Willebrand factor (vWF) multimers, leading to thrombus disintegration.17 Even the JAK2V617F mutation has been shown to induce proteolysis of vWF multimers,25 although we observed increased pulmonary thrombosis in mice engrafted with JAK2V617F cells. Possible antithrombotic actions of NAC may be due the reduction of vWF multimers29 as well its interactions with other clotting factors containing disulfide bonds. Moreover, NAC has been found to downregulate lysyl oxidase (LOX) activity through glutathione replenishment in idiopathic pulmonary fibrosis model.30 Interestingly, overexpression of LOX has been identified in MPN, and LOX overexpression in platelets leads to a prothrombotic phenotype that increases platelet adhesion to collagen.31
In summary, this work identifies NAC as a potential agent to reduce thrombosis in MPN. The exact mechanism by which NAC rescues JAK2V617F from thrombotic death requires further investigation, although our findings suggest that NAC impacts the interaction between platelets and neutrophils in MPN. Given that NAC is a low-cost, widely available, safe agent, our results encourage the evaluation of NAC in patients with MPN for reduction of thrombotic risk. Thus, we are currently developing a clinical trial investigating the impact of NAC in MPN. Moreover, NAC may also prove to be beneficial in other clonal hematologic conditions such as clonal hematopoiesis of indeterminate potential,32 which is associated with coronary heart disease as well as thrombosis.
Acknowledgments
The authors thank Eric Pearlman and Martin Minns (both from the University of California, Irvine) for their helpful feedback on NETs and providing protocols and reagents for NET quantification.
This work was supported by an MPN Research Foundation challenge grant (A.G.F.), a Department of Defense Career Development Award (A.G.F.), and University of California Cancer Research Coordinating Committee (A.G.F.). Research reported in this publication was supported by the National Institutes of Health, National Cancer Institute through grant T32CA009054 (B.M.C. and G.R.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: B.M.C. performed experiments, analyzed the data, made the figures, and wrote the manuscript; G.R. performed the experiments, designed experiments, and wrote the manuscript; S.H. and X.C. performed platelet activation experiments; L.F.M.L performed NET experiments; S.B. and H.Y.L. discussed results and provided comments and feedback; A.G.F. planned, designed, and coordinated the research and wrote the manuscript; and all authors read the manuscript and approved its content.
Conflict-of-interest disclosure: A.G.F. is a member of the Incyte and Celgene speakers bureaus. The remaining authors declare no competing financial interests.
Correspondence: Angela G. Fleischman, Division of Hematology/Oncology, Department of Medicine, University of California, Irvine, 839 Health Sciences Rd, Irvine, CA 92697; e-mail: agf@uci.edu.

