

Key Points
Findings support use of PB samples for chronic myeloid neoplasms and for acute leukemias with sufficient circulating disease.
In acute leukemias, BM appears to be superior to PB for monitoring measurable residual disease, even in the absence of BM excess blasts.
Introduction
The classification, risk stratification, and treatment selection of patients with hematologic malignancies are heavily influenced by recurrent molecular alterations determined by next-generation sequencing (NGS).1-5 Peripheral blood (PB) and bone marrow (BM) are generally considered equivalent sample sources, especially if similarly involved by disease, but large studies comparing the mutational profiles of paired PB and BM samples (ie, taken from the same patient at the same time point of their disease history) have not been performed.6,7 We report a retrospective analysis of 164 patients from a single institution with suspected or known hematologic diseases for whom both PB and BM NGS was performed.
Methods
Sequencing was performed at Brigham and Women’s Hospital (Boston, MA), using the 95-gene Rapid Heme Panel (RHP) amplicon NGS assay as previously published (see also supplemental Methods).6 All RHP assays performed between September 2014 and November 2017 were included in this institutional review board–approved study. After that time, stricter practice guidelines limited concurrent testing. A total of 2636 RHP assays were run on BM samples obtained from 1226 patients, and 1371 RHP assays were performed on PB samples obtained from 985 patients. Paired RHP assays, defined as a PB and BM sample submitted for NGS within 14 days or less of each other to minimize discordance because of therapy, were identified for 164 patients (n = 85 male, n = 79 female; mean age at RHP = 57 years; range, 14-92 females and 3-87 males). For patients with multiple paired samples, only the first pair was included. Diagnoses of all patients with paired samples and treatment between samples were confirmed, and all variants were manually retiered by a single pathologist for internal consistency. All pathogenic/presumed pathogenic variants were considered for this study. Manual review of the binary alignment map (BAM) files for all discordant variants was performed. Methylcellulose colony-forming studies were conducted for 3 unpaired samples (1 PB, 2 BM) with a concurrent or recent NGS result (reference for variants) and persistent disease. After 9 days in culture, individual colonies were selected, and DNA was extracted. Each known targeted locus was amplified by polymerase chain reaction and submitted for Sanger sequencing of the mutation loci. Reference variant allele fractions (VAFs) ranged from 2.9% to 92.6%.
Results and discussion
For 73% of patients (120 of 164), PB was submitted before BM, and 51% (83 of 164) of samples were submitted within 3 days of each other (supplemental Figure 1A). Most patients (91%, 149 of 164) were not treated between samples. Supportive treatment was started before the first RHP for 5 patients. Anti-leukemia treatment was initiated before the second RHP for 7 patients. Records were unavailable for 3 patients. Most patients presented with a suspicion or known diagnosis of a myeloid stem cell disorder (n = 129, 79%), with fewer lymphoid neoplasms (n = 32, 20%) and 3 (1.8%) mixed lineage acute leukemias (MPALs; specific diagnostic subcategories are listed in supplemental Table 1). For 46 of 164 patients with paired samples (28%), no pathogenic variants were called in either sample. The same pathogenic variants were called in both paired samples for 84 of 164 patients (51%), with a total of 207 concordant variants: 17 concordant variants (average, 1.6 per patient; range, 1-4) in 11 patients with lymphoid or MPAL malignancies and 190 concordant variants (average, 2.6 per patient; range, 1-8) in 73 patients with myeloid diagnoses. At least 1 discordant pathogenic variant (ie, present in 1 sample but not the other) was initially reported for 34 of 164 patients (21%). Variant distribution for all patients with paired samples according to diagnosis is depicted in supplemental Figure 1B. Based solely on the initially reported variants, there were 278 concordant and 51 discordant pathogenic variants (supplemental Table 2) and 4370 presumed concordant negative calls (number of negative samples multiplied by the number of genes in panel), for a 98.9% concordance between PB and BM overall and a sensitivity of 88.0%, specificity of 99.7%, positive predictive value (PPV) of 95.5%, and negative predictive value (NPV) of 99.1% for PB compared with BM (supplemental Table 3).
We next interrogated all 34 patients with discordant variants per NGS report. Disease characteristics, prior treatment, and hematologic parameters are summarized in supplemental Table 4. Of the 34 patients, 33 received no therapy between sample submissions; 1 patient underwent ongoing hydroxyurea treatment at the time of both samples. The mean number of called variants was higher in BM (n = 3.2; range, 1-10) than in PB (n = 2.5; range, 0-9). In these 34 patients, a total of 71 concordant variants were identified in both PB and BM, whereas 51 variants were discordant (38 found only in BM, 13 found only in PB). In more than 50% of cases (28 of 51 variants in 18 of 34 patients), the discordant variants were not directly actionable (supplemental Table 5). In addition, 4 variants (from 4 patients) potentially indicated clonal hematopoiesis, whereas 5 variants (from 5 patients) indicated poor prognosis in acute myeloid leukemia (AML) or myelodysplastic syndromes (in TP53 and RUNX1); these variants were found only in the BM. Variants in NOTCH1, IL7R, and JAK3 were considered potentially targetable in 3 acute lymphoblastic leukemia (ALL) patients and were identified only in BM. Last, 10 targetable mutations in FLT3, IDH1, and MYD88 were identified in 8 patients in BM only. These 8 patients were either new diagnoses of AML (n = 4) or Waldenström macroglobulinemia (n = 1) or cases of measurable residual disease (MRD) in AML (n = 3).
Manual review of the BAM files for all discordant variants revealed that most of the uniquely called variants (36 of the 45 discordant variants, 80%) were also identified in the second sample (supplemental Figure 2) but were not originally reported because of low VAFs (<5%, including discrepancies in tumor burden between the compartments, especially for the lymphoid neoplasms) and lower or poor coverage (Figure 1A-B). Six variants were excluded from further analysis for the following reasons: 3 FLT3-ITDs because of the nature of the custom FLT3-ITD caller making it impossible to conduct a manual review of the BAM files, 2 variants in 1 patient because of irretrievable BAM files, and 1 variant where there was no coverage at all at the locus in question. However, a total of 9 variants found in 7 patients remained truly discordant (Figure 1C), for a final concordance of 99.8% with 98.4% sensitivity, 99.9% specificity, 98.7% PPV, and 99.9% NPV of PB compared with BM (supplemental Table 6). A complete list of all discordant variants, including VAFs and coverage, can be found in supplemental Table 5.
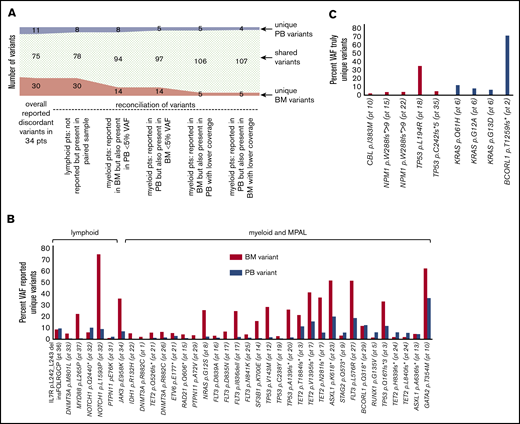
A total of 116 reported pathogenic variants available for manual review in paired samples from 34 patients. (A) Overview of reasons for variants not having been called in the paired sample and resulting changes in numbers of unique and shared variants. Six variants from 5 patients where presence could not be verified in the other sample because of lack of coverage, custom FLT3-ITD caller, and irretrievable BAM files are excluded. (B) VAFs of reported unique variants, grouped by lymphoid vs myeloid/ MPAL diagnoses and pathways. (C) VAFs of truly unique variants. All variants and VAFs were confirmed by manual review of BAM files.
A total of 116 reported pathogenic variants available for manual review in paired samples from 34 patients. (A) Overview of reasons for variants not having been called in the paired sample and resulting changes in numbers of unique and shared variants. Six variants from 5 patients where presence could not be verified in the other sample because of lack of coverage, custom FLT3-ITD caller, and irretrievable BAM files are excluded. (B) VAFs of reported unique variants, grouped by lymphoid vs myeloid/ MPAL diagnoses and pathways. (C) VAFs of truly unique variants. All variants and VAFs were confirmed by manual review of BAM files.
Variants might be discordant between samples if they are found preferentially only in undifferentiated cells (blasts) or alternatively only in mature cells. The unique BM variants included CBL p.I383M, TP53 p.L194R, and NPM1 p.W288fs*>9 (unique in 2 different patients) in myeloid patients and TP53 p.C242fs*5 in 1 patient with B-cell ALL. Four of the 5 of the BM-only variants involved mutations in NPM1 and TP53, known leukemia driver events. These 4 examples were cases of AML and ALL where patients had no circulating blasts (patients 15, 22, 18, and 35), likely explaining the absence of these leukemia drivers in PB. These results indicate the importance of using BM for monitoring MRD, even when the BM does not have excess blasts (patients 22 and 35 had less than 5% BM blasts).
PB-only findings raised the possibility that these mutations might be found only in terminally differentiated circulating cells. Unique PB variants included 3 KRAS variants in 1 patient and a BCORL1 variant. Excluding the BCORL1 variant that was likely missed in PB because of low coverage (only 44×), the unique PB variants involved signal transduction pathways that are typically subclonal progression mutations.1 Colony-forming assays were performed on 3 new samples acquired prospectively on the basis of historically having signal transduction pathway mutations (Figure 2). In each of these samples, at least 1 signal transduction pathway mutation was found in 100% of colonies, suggesting that signal transduction variants in the RAS pathway are not present fundamentally only in more differentiated cells. Other low level subclonal variants (as determined by the reference historical or concurrent NGS study) were likely not uniformly found in colonies for stochastic reasons, given the low numbers of colonies assessed (6, 10, and 10 colonies, respectively). This is in stark contrast to the p.D816V variant KIT, where the cells with KIT mutations in most patient samples lack colony-forming ability.8
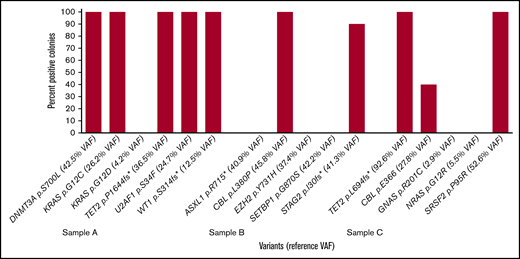
Percentage of colonies positive for variant in methylcellulose colony-forming assays on 3 samples with reference variants determined from a concurrent RHP on a separate specimen. PB methylcellulose colony-forming assays were performed on 3 samples with reference variants determined from a concurrent peripheral blood specimen (sample A) or concurrent or recent BM sample (samples B and C). Six colonies were obtained for sample A, and 10 colonies were obtained for samples B and C after 9 days in culture.
Percentage of colonies positive for variant in methylcellulose colony-forming assays on 3 samples with reference variants determined from a concurrent RHP on a separate specimen. PB methylcellulose colony-forming assays were performed on 3 samples with reference variants determined from a concurrent peripheral blood specimen (sample A) or concurrent or recent BM sample (samples B and C). Six colonies were obtained for sample A, and 10 colonies were obtained for samples B and C after 9 days in culture.
In summary, retrospective analysis of paired PB and BM samples submitted for clinical NGS testing with a diagnosis or concern for a hematologic malignancy revealed an initial 98.9% concordance, with 88% sensitivity of PB in identifying all BM variants (99.7% specificity, 95.5% PPV, and 99.1% NPV). Despite the absence of some mutations in the PB, confirmation of a clonal hematopoietic process would have been correctly identified in PB in 153 of 164 paired samples (93%), supporting the use of PB, especially in the diagnostic context when PB is involved (chronic myeloid neoplasms, circulating chronic lymphoid neoplasms, and acute leukemias with sufficient circulating blasts).9 Manual review revealed that most discrepancies were caused by low-level subclonal events and areas with poor coverage, and only 2.8% of variants (9 of 323) were truly discrepant, emphasizing the importance of reporting undercovered regions and providing a limit of detection. Variable detection at the lower limit of detection or coverage is a stochastic problem encountered by all assays. Although the number of cases was too small to reach definitive assessment of mutational categories affected by the discrepancies, PB-only variants were enriched for signal transduction pathway variants, but colony-forming assays determined that these variants are not found solely in terminally differentiated cells. By contrast, BM-only variants were enriched for known leukemia drivers and support the recommendation of using BM samples for monitoring MRD and at diagnosis of an acute leukemia. These findings should be confirmed in larger multi-institutional studies.
To request data, please contact the corresponding author at askim@bwh.harvard.edu.
Acknowledgment
The authors thank Frank C. Kuo in memoriam for spearheading the development of the assay used in this study.
Authorship
Contribution: F.L. and P.D.M. reviewed NGS and clinical notes, performed manual review of BAM files, analyzed the data, and wrote and edited the manuscript; F.L. prepared figures; D.W. performed colony formation assay experiments, analyzed data, and edited the manuscript; A.S.K. designed and provided the conceptual framework of the study, performed NGS analyses, reviewed variants and BAM files and pathology diagnoses, analyzed the data, wrote and edited the manuscript, and prepared figures; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: A.S.K. has received consulting fees from LabCorp, Inc. The remaining authors declare no competing financial interests.
Correspondence: Annette S. Kim, Brigham and Women's Hospital, Pathology, 75 Francis St, Thorn 613A, Boston, MA 02115; e-mail: askim@bwh.harvard.edu.

