

Key Points
Ibrutinib added to 10-day decitabine does not improve response or survival in newly diagnosed AML patients unsuitable for intensive chemotherapy.
Mutated TP53 correlates with high response and STAG2/IDH2/ASXL1 with low response rates.

Abstract
The treatment of older, unfit patients with acute myeloid leukemia (AML) is challenging. Based on preclinical data of Bruton tyrosine kinase expression/phosphorylation and ibrutinib cytotoxicity in AML blasts, we conducted a randomized phase 2 multicenter study to assess the tolerability and efficacy of the addition of ibrutinib to 10-day decitabine in unfit (ie, Hematopoietic Cell Transplantation Comorbidity Index ≥3) AML patients and higher risk myelodysplasia patients (HOVON135/SAKK30/15 trial). In total, 144 eligible patients were randomly (1:1) assigned to either 10-day decitabine combined with ibrutinib (560 mg; sequentially given, starting the day after the last dose of decitabine) (n = 72) or to 10-day decitabine (n = 72). The addition of ibrutinib was well tolerated, and the number of adverse events was comparable for both arms. In the decitabine plus ibrutinib arm, 41% reached complete remission/complete remission with incomplete hematologic recovery (CR/CRi), the median overall survival (OS) was 11 months, and 2-year OS was 27%; these findings compared with 50% CR/CRi, median OS of 11.5 months, and 2-year OS of 21% for the decitabine group (not significant). Extensive molecular profiling at diagnosis revealed that patients with STAG2, IDH2, and ASXL1 mutations had significantly lower CR/CRi rates, whereas patients with mutations in TP53 had significantly higher CR/CRi rates. Furthermore, multicolor flow cytometry revealed that after 3 cycles of treatment, 28 (49%) of 57 patients with available bone marrow samples had no measurable residual disease. In this limited number of cases, measurable residual disease revealed no apparent impact on event-free survival and OS. In conclusion, the addition of ibrutinib does not improve the therapeutic efficacy of decitabine. This trial was registered at the Netherlands Trial Register (NL5751 [NTR6017]) and has EudraCT number 2015-002855-85.
Introduction
About 75% of patients with acute myeloid leukemia (AML) are aged ≥60 years.1 The optimal treatment of older AML patients in daily clinical practice remains challenging and is dependent on patient characteristics (ie, age, performance, comorbidities), disease characteristics (ie, cytogenetic and molecular abnormalities, white blood cell [WBC] count), and the preference of the patient.2 The hypomethylating agents (HMAs) azacitidine and decitabine have relatively mild side effects and are particularly feasible for the treatment of older patients and patients with comorbidities. Although not in their primary analyses, recent phase 3 trials have shown the superiority of azacitidine and decitabine compared with conventional care for older patients with AML in a prespecified analysis (censoring patients who received AML treatment after discontinuing study drug [azacitidine]) or after an unplanned analysis (after more events [decitabine]).3-5 Results of treatment with modifications of the standard azacitidine (7 days, 75 mg/m2 subcutaneously; every 4 weeks) and decitabine (5 days, 20 mg/m2 IV; every 4 weeks) schedules have been reported. Initial studies evaluating the 10-day decitabine schedule have shown promising results. In a hallmark study, 52 older patients newly diagnosed with AML were treated with the 10-day decitabine schedule; 21 patients (40%) achieved a complete remission (CR), and the median overall survival (OS) was 318 days.6 Various retrospective analyses have confirmed these favorable data.7,8 A phase 3 trial (#NCT02172872) in older patients with AML (aged ≥60 years) that compares conventional intensive chemotherapy based on cytarabine combined with an anthracycline (“3 + 7”) with the 10-day decitabine schedule, followed by allogeneic hematopoietic cell transplantation, is currently in progress.
It remains an important clinical challenge to further improve the cumulative CR/complete remission with incomplete hematologic recovery (CR/CRi) rates and survival of the 10-day decitabine schedule. Based on preclinical scientific evidence, we hypothesized that ibrutinib, an oral inhibitor of Bruton tyrosine kinase (BTK) that is approved by the US Food and Drug Administration for the treatment of mantle cell lymphoma, chronic lymphocytic leukemia, and Waldenström macroglobulinemia, could be an interesting new drug to add to decitabine.9 Although BTK is critical to the B-cell receptor signaling cascade, AML cell lines also have high expression of BTK, comparable to B-cell lines (www.broadinstitute.org/ccle). It has been reported that BTK is phosphorylated in the leukemic blasts in >90% of AML patient samples but not in healthy CD34+ cells.10-12 Furthermore, it seems that ibrutinib (alone or combined with cytarabine or daunorubicin) inhibits blast proliferation from human AML in vitro.10 The in vitro cytotoxicity of ibrutinib in AML is comparable to the in vitro cytotoxicity of primary chronic lymphocytic leukemia cells (ie, 50% inhibitory concentration of ∼5 µM). BTK-targeted RNA interference knockdown reduces colony-forming capacity of primary AML blasts and proliferation of AML cell lines. Silencing of BTK inhibits protein kinase B (AKT) and extracellular signal-regulated kinase (ERK) pathways in AML cells. It has been shown that ibrutinib’s antiproliferative effects in AML are mediated via an inhibitory effect on downstream NF-κB survival pathways.11 Moreover, ibrutinib seems to inhibit AML cell adhesion to bone marrow stroma.13 These experimental results provide a biological rationale for the clinical evaluation of BTK inhibition in patients with AML.
Here we present the final analysis of the HOVON135/SAKK-30/15 study. In this phase 2 study, older patients (aged ≥66 years) with AML or higher risk myelodysplasia (MDS) (Revised International Prognostic Scoring System score >4.5) considered not suitable for intensive cytotoxic treatment were randomized to receive 10-day decitabine with or without ibrutinib. The aim was to assess the value of adding ibrutinib to 10-day decitabine with respect to CR/CRi after 3 cycles of treatment (primary end point) and OS (secondary end point).
Methods
Study design, patients, and treatment
This was a randomized phase 2 multicenter study (HOVON135; SAKK30/15; EudraCT #2015-002855-85) (protocol is available at: www.hovon.nl). Previously untreated adults who were ≥66 years old and not considered eligible for intensive chemotherapy, with a cytopathologically confirmed diagnosis of AML or with higher risk MDS (Revised International Prognostic Scoring System score >4.5), a World Health Organization (WHO) performance status ≤2, and written informed consent, were eligible. Patients were considered not suitable for intensive chemotherapy if they had comorbidities that would put them at high risk for early death when treated with intensive chemotherapy (defined as hematopoietic cell transplantation comorbidity index [HCT-CI] ≥3) or if the patient declined to receive intensive chemotherapy. The limit for HCT-CI ≥3 was chosen because this value has been shown to be associated with an early mortality rate (<30 days) of ∼30% in older patients with AML who are treated with intensive chemotherapy.14 Based on the karyotype and molecular genotype of the leukemic cells, patients were classified into prognostic categories according to the European LeukemiaNet (ELN) 2017 criteria.15
Patients were randomly assigned (1:1) to 10-day decitabine without (arm A) or with (arm B) ibrutinib 560 mg (Figure 1). In arm B, ibrutinib was given sequentially with decitabine, starting the day after the last dose of decitabine until the day before the start of the next cycle of decitabine. (Decitabine and ibrutinib were provided free of charge by Janssen Pharmaceutics.) The duration of decitabine in the second and third cycle (ie, 5 or 10 days) was dependent on the percentage of bone marrow blasts (ie, <5% or ≥5%), as determined by cytomorphology, at the day 28 bone marrow evaluation. Cycle 2 started within 1 week after the day 28 bone marrow evaluation, independent of recovery of peripheral counts. Cycle 3 started if neutrophils were ≥0.5 × 109/L or platelets were ≥25 × 109/L, with a maximum delay of 2 weeks. From cycle 4, treatment could only be continued with 5-day decitabine (with or without ibrutinib).
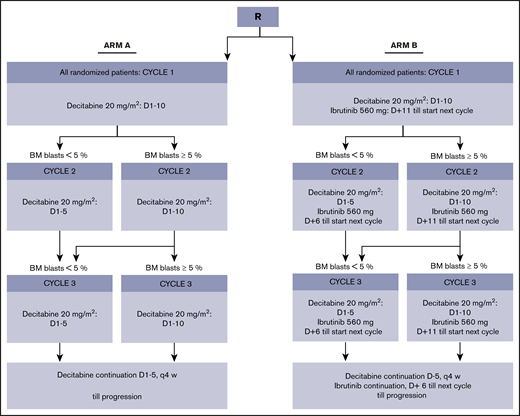
Molecular analyses and flow cytometry
Bone marrow aspirations or peripheral blood samples at diagnosis were taken. Blasts and mononuclear cells at diagnosis were purified by Ficoll-Hypaque (Nygaard, Oslo, Norway) density gradient centrifugation and cryopreserved. After thawing, cells were lysed in RLT solution with the addition of dithiothreitol (Qiagen, Venlo, The Netherlands). High-quality DNA and RNA were extracted by using QIAsymphony (Qiagen). DNA concentration was measured by Qubit Fluorometric Quantitation (Thermo Fisher Scientific, Wilmington, DE) and RNA concentration using the Implen NanoPhotometer (Westburg, Leusden, The Netherlands). CBFB-MYH11, RUNX1-RUNX1T1, and FLT3 internal tandem duplication mutations were determined as previously described.16,17 Mutations in 38 of 54 genes frequently mutated in hematologic malignancies were determined by targeted next-generation sequencing (NGS) with the TruSight Myeloid Sequencing panel, following the manufacturer’s protocol (Illumina, San Diego, CA). The NGS libraries were paired-end sequenced (2 × 221 bp) on a MiSeq System (Illumina). Variants were called as described.18
Residual disease detection by using multiparameter flow cytometry was performed as previously described.19 The residual disease percentage was defined as the number of leukemia-associated immunophenotype (LAIP) cells within the bone marrow compartment. The threshold between residual and no residual disease based on flow cytometry was established and validated on 0.1%.19,20 Information on clones and commercial sources of all monoclonal antibodies used is provided in supplemental Table 1.
Study end points and statistical analysis
The aim of this study was to decide whether the addition of ibrutinib to standard induction treatment could be sufficiently effective to warrant continuation to a phase 3 study. The observed difference in CR/CRi rates between investigational arm B and standard arm A was used as the criterion in decision rules. At final analysis, the decision to stop because of inefficacy (ie, not to continue with phase 3) was based on observed difference in CR/CRi rates ≤0 or if the upper limit of the 80% confidence interval (CI) was <25%.
The primary end point of the study was cumulative CR/CRi after 3 cycles (for all patients [MDS and AML]). A patient was considered to have a CR/CRi if this end point was attained within the first 3 induction cycles and the patient did not relapse on a consecutive cycle (within these 3 induction cycles). All other patients without “CR/CRi after 3 cycles” (including partial remission and morphologic leukemia-free state [MLFS]) were classified as “induction failures.”
Secondary end points included safety, tolerability, and efficacy profile (response rate [CR, CRi, MLFS, partial remission]), event-free survival (EFS), and OS. OS is defined as the time from the date of randomization to the date of death, whatever the cause. The follow-up of patients still alive was censored at the moment of last visit/contact. EFS was defined as the time from registration to induction failure, death, or relapse, whichever occurred first. Kaplan-Meier survival curves and Cox regression tests were used to compare the survival distributions between the treatment arms. For statistical comparison of mutation frequencies and clonal characteristics between individuals with and without a response, Fisher’s exact test and Mann-Whitney U tests were used.
Results
Study patients and treatment
From 1 September 2016 until 13 June 2018, a total of 144 patients were registered for the study. The database was closed on 17 February 2020. The median follow-up for all patients is 11.3 months, and the median follow-up for patients still alive is 23.5 months. Patient characteristics are described in Table 1. The main patient-related factors (sex, age, performance, and comorbidities) and disease-related factors (2017 ELN risk, secondary AML, MDS, and WBC count) were equally distributed over both arms. In the decitabine arm (arm A), 65% were male, median age was 76 years (range, 68-84 years), 89% had performance score WHO 0 to 1, and 42% had an HCT-CI ≥3. In the decitabine plus ibrutinib arm (arm B), 58% were male, median age was 75 years (range, 66-89 years), 84% had performance score WHO 0 to 1, and 42% had an HCT-CI ≥3. In arm A, 47% had 2017 ELN adverse risk and 18% secondary AML; in arm B, 53% had 2017 ELN adverse risk and 21% secondary AML.
Patient characteristics
| Characteristic | 10-d decitabine (n = 72) | 10-d decitabine + ibrutinib (n = 72) |
|---|---|---|
| Sex, male/female | 47/25 (65/35) | 42/30 (58/42) |
| Age, median (range), y | 76 (68-84) | 75 (66-89) |
| WHO performance | ||
| WHO 0 | 26 (36) | 25 (35) |
| WHO 1 | 38 (53) | 35 (49) |
| WHO 2 | 8 (11) | 12 (17) |
| Comorbidity | ||
| HCT-CI 0-2 | 42 (58) | 42 (58) |
| HCT-CI ≥3 | 30 (42) | 30 (42) |
| ELN risk group (AML) | ||
| Favorable | 15 (21) | 10 (14) |
| Intermediate | 12 (17) | 11 (15) |
| Adverse | 34 (47) | 37 (51) |
| Unknown | 0 (0) | 2 (3) |
| Primary AML | 48 (67) | 45 (63) |
| Secondary AML | 13 (18) | 15 (21) |
| MDS | 11 (15) | 12 (17) |
| WBC, median (range), ×109/L | 2.95 (0.70-29.9) | 3.20 (0.60-30.0) |
| Characteristic | 10-d decitabine (n = 72) | 10-d decitabine + ibrutinib (n = 72) |
|---|---|---|
| Sex, male/female | 47/25 (65/35) | 42/30 (58/42) |
| Age, median (range), y | 76 (68-84) | 75 (66-89) |
| WHO performance | ||
| WHO 0 | 26 (36) | 25 (35) |
| WHO 1 | 38 (53) | 35 (49) |
| WHO 2 | 8 (11) | 12 (17) |
| Comorbidity | ||
| HCT-CI 0-2 | 42 (58) | 42 (58) |
| HCT-CI ≥3 | 30 (42) | 30 (42) |
| ELN risk group (AML) | ||
| Favorable | 15 (21) | 10 (14) |
| Intermediate | 12 (17) | 11 (15) |
| Adverse | 34 (47) | 37 (51) |
| Unknown | 0 (0) | 2 (3) |
| Primary AML | 48 (67) | 45 (63) |
| Secondary AML | 13 (18) | 15 (21) |
| MDS | 11 (15) | 12 (17) |
| WBC, median (range), ×109/L | 2.95 (0.70-29.9) | 3.20 (0.60-30.0) |
Data are n (%) except as noted.
All 72 patients allocated to arm A started 10-day decitabine; 59 patients (82%) received a second cycle, and 48 patients (67%) received a third cycle. In arm B, 2 patients (of 72) went off protocol before the start of the first 10-day decitabine cycle; 51 patients (82%) received a second cycle, and 39 patients (54%) received a third cycle. Main reasons to discontinue treatment were death, adverse events (AEs) preventing further treatment, and progressive disease/relapse (Figure 2). “Full-dose” decitabine (including “delayed”) was given in arm A in 99%, 97%, and 98% and in arm B in 100%, 92%, and 92% in the first 3 cycles. The full-dose ibrutinib was given in 53%, 45%, and 38% during the first 3 cycles; “dose reduced >10%” and/or “delayed” in 38%, 33%, and 44% during the first 3 cycles; “interrupted and resumed” in 6%, 12%, and 5% during the first 3 cycles; and “not given” in 3%, 10%, and 13% during the first 3 cycles. The relatively high number of patients whose dose was “reduced by >10%” and/or “delayed” might be explained by the study protocol, which required reducing the dose of ibrutinib from 560 mg to 140 mg in case moderate or strong cytochrome P450 3A inhibitors were used (eg, ciprofloxacin, posaconazole, voriconazole). The median number of decitabine cycles applied was 4 in arm A (range, 1-26) and 3 (range, 1-29) in arm B. Three patients received an allogeneic hematopoietic cell transplantation (1 in arm A, 2 in arm B).

CONSORT study diagram. Arm A = 10-day decitabine; arm B = 10-day decitabine plus ibrutinib. PD, progressive disease; Tx, treatment.
CONSORT study diagram. Arm A = 10-day decitabine; arm B = 10-day decitabine plus ibrutinib. PD, progressive disease; Tx, treatment.
Treatment outcome according to randomization
The primary end point of this study was the cumulative CR/CRi rate during the first 3 cycles, which was attained in 29 of 72 patients (40%; 80% CI, 33-48) in arm A and 22 of 72 patients (31%; 80% CI, 24-38) in arm B. Thus, there is no difference in CR/CRi between arms B and A. Some patients obtained a CR/CRi after the first 3 cycles (the primary end point), resulting in a total CR/CRi rate (on protocol) of 50% in arm A and 41% in arm B.
At time of the final analysis, a total of 116 patients had died. Median OS was 11.5 months in arm A and 11.0 months in arm B. OS at 12 and 24 months was similar between arms (A vs B): 47% (95% CI, 35-58) vs 49% (95% CI, 37-60) and 21% (95% CI, 12-31) vs 27% (95% CI, 17-38), respectively (log-rank test, P = .48) (Figure 3).

Kaplan-Meier estimates for OS. Arm A = 10-day decitabine arm; arm B = 10-day decitabine plus ibrutinib. Median OS was almost 12 months in both arms.
Kaplan-Meier estimates for OS. Arm A = 10-day decitabine arm; arm B = 10-day decitabine plus ibrutinib. Median OS was almost 12 months in both arms.
The CR/CRi rates within the first 3 cycles were 40% for favorable, 35% for intermediate, and 35% for adverse risk AML according to ELN 2017 criteria, and 35% for the higher risk MDS patients. The OS was not different for the favorable, intermediate, and adverse risk AML and higher risk MDS subgroups (log-rank test, P = .08) (supplemental Figure 1).
Tolerability of treatment
During cycle 1, the median number of nights in the hospital was 13 (range, 0-42) in arm A and 17 (range, 0-56) in arm B; during cycle 2, the median number of nights in the hospital was 9 (range, 0-32) in arm A and 10 (range, 0-33) in arm B (Table 2). During the third cycle, the median number of nights in the hospital for both arms was 0. The median number of red blood cell transfusions in arms A and B were comparable: 5, 4, and 2, respectively, during cycles 1, 2, and 3. The median number of platelet transfusions in arms A and B was also comparable: 3, 1, and 0 during cycles 1, 2, and 3.
Tolerability of treatment per treatment arm
| Variable | 10-d decitabine (n = 72) | 10-d decitabine + ibrutinib (n = 72) |
|---|---|---|
| Cycle 1, median | ||
| Nights in hospital | 13 | 17 |
| No. of RBC transfusions | 5 | 5 |
| No. of platelet transfusions | 3 | 3 |
| Cycle 2, median | ||
| Nights in hospital | 9 | 10 |
| No. of RBC transfusions | 4 | 4 |
| No. of platelet transfusions | 1 | 1 |
| Cycle 3, median | ||
| Nights in hospital | 0 | 0 |
| No. of RBC transfusions | 2 | 2 |
| No. of platelet transfusions | 0 | 0 |
| AEs, any, n (%) | ||
| Grade 2 | 50 (9) | 58 (11) |
| Grade 3 | 81 (14) | 92 (18) |
| Grade 4 | 30 (5) | 21 (4) |
| Patients with SAEs, n (%) | ||
| 0 SAE | 14 (19) | 9 (13) |
| 1 SAE | 25 (35) | 32 (44) |
| 2 SAEs | 12 (17) | 21 (29) |
| 3 SAEs | 13 (18) | 6 (8) |
| >3 SAEs | 8 (11) | 4 (6) |
| Variable | 10-d decitabine (n = 72) | 10-d decitabine + ibrutinib (n = 72) |
|---|---|---|
| Cycle 1, median | ||
| Nights in hospital | 13 | 17 |
| No. of RBC transfusions | 5 | 5 |
| No. of platelet transfusions | 3 | 3 |
| Cycle 2, median | ||
| Nights in hospital | 9 | 10 |
| No. of RBC transfusions | 4 | 4 |
| No. of platelet transfusions | 1 | 1 |
| Cycle 3, median | ||
| Nights in hospital | 0 | 0 |
| No. of RBC transfusions | 2 | 2 |
| No. of platelet transfusions | 0 | 0 |
| AEs, any, n (%) | ||
| Grade 2 | 50 (9) | 58 (11) |
| Grade 3 | 81 (14) | 92 (18) |
| Grade 4 | 30 (5) | 21 (4) |
| Patients with SAEs, n (%) | ||
| 0 SAE | 14 (19) | 9 (13) |
| 1 SAE | 25 (35) | 32 (44) |
| 2 SAEs | 12 (17) | 21 (29) |
| 3 SAEs | 13 (18) | 6 (8) |
| >3 SAEs | 8 (11) | 4 (6) |
RBC, red blood cell; SAE, serious AE.
The total number of grade 2 to 4 AEs was 577 (72 of which were serious AEs) in arm A and 524 (72 of which were serious AEs) in arm B (supplemental Table 2). The number of any Common Terminology Criteria for Adverse Events grade 2, 3, or 4 AEs was 9%, 14%, and 5% in arm A and 11%, 18%, and 4% in arm B, respectively. Grade 3/4 infections and infestations were 9% in both arms. The early death rate (within 30 days from registration) was 13% in arm A and 3% in arm B.
Treatment outcome according to molecular profile
Comprehensive molecular analysis at diagnosis was performed in 140 cases; 7 (5%) of these had no detectable gene mutations. The most frequently mutated genes were DNMT3A (33%), TET2 (26%), ASXL1 (25%), SRSF2 (24%), RUNX1 (23%), and TP53 (19%) (Table 3). The typical molecular abnormalities in (younger) AML, mutated NPM1 and FLT3 internal tandem duplication, were both observed in only 14% of these older patients. The various mutations were generally equally distributed over both treatment groups (supplemental Figure 3A).
Molecular diagnostics and response
| Gene | No. of individuals with mutations | No. (%) CR/CRi after 3 cycles |
|---|---|---|
| DNMT3A | 46 | 18 (39) |
| TET2 | 36 | 10 (28) |
| ASXL1 | 35 | 7 (20) |
| SRSF2 | 34 | 8 (24) |
| RUNX1 | 32 | 9 (28) |
| TP53 | 27 | 15 (56) |
| IDH2 | 23 | 4 (17) |
| NRAS | 21 | 8 (38) |
| NPM1 | 19 | 5 (26) |
| STAG2 | 17 | 2 (12) |
| U2AF1 | 15 | 8 (53) |
| SF3B1 | 15 | 6 (40) |
| JAK2 | 12 | 6 (50) |
| BCOR | 12 | 5 (42) |
| EZH2 | 11 | 2 (18) |
| CEBPA | 10 | 2 (20) |
| FLT3 | 10 | 4 (40) |
| Gene | No. of individuals with mutations | No. (%) CR/CRi after 3 cycles |
|---|---|---|
| DNMT3A | 46 | 18 (39) |
| TET2 | 36 | 10 (28) |
| ASXL1 | 35 | 7 (20) |
| SRSF2 | 34 | 8 (24) |
| RUNX1 | 32 | 9 (28) |
| TP53 | 27 | 15 (56) |
| IDH2 | 23 | 4 (17) |
| NRAS | 21 | 8 (38) |
| NPM1 | 19 | 5 (26) |
| STAG2 | 17 | 2 (12) |
| U2AF1 | 15 | 8 (53) |
| SF3B1 | 15 | 6 (40) |
| JAK2 | 12 | 6 (50) |
| BCOR | 12 | 5 (42) |
| EZH2 | 11 | 2 (18) |
| CEBPA | 10 | 2 (20) |
| FLT3 | 10 | 4 (40) |
Data from 140 patients with available extensive molecular analyses are considered.
The response rates (CR/CRi) after 3 cycles of treatment were not different for most of the molecular abnormalities (supplemental Figure 3B). The response rates (CR/CRi within 3 cycles) for the most frequently mutated genes in the evaluable 140 patients are depicted in Table 3. Of those mutations that were observed at least 10 times, only mutations in TP53 were associated with a significantly higher CR/CRi rate (56%) during the first 3 cycles (Figure 4A). This favorable CR/CRi rate did not translate into superior OS of patients with mutated TP53 (supplemental Figure 4). Indeed, the inferior OS of patients with mutated TP53 was borderline significant compared with those without mutated TP53 (P = .05). Among the mutations that were observed at least 10 times, patients with mutations in STAG2, IDH2, and ASXL1 showed significantly reduced CR/CRi rates within 3 cycles of decitabine (12% for STAG2, 17% for IDH2, and 20% for ASXL1). Although numbers were low, the reduced CR/CRi rates for these genes did not result in inferior OS of patients with these genes mutated (supplemental Figure 5).

Associations between molecular and cytogenetic abnormalities (with prevalence ≥10) and response. (A) The association between individual frequently mutated genes (≥10 times) and frequently observed cytogenetic abnormalities (≥10 times) and response (CR/CRi) after 3 cycles. Odds ratios are displayed on a log10 scale, with lines extending to the 95% CI. Horizontal bars indicate the proportion with a mutation in the respective gene for patients with (blue) and without (red) response, with absolute numbers given for each gene. Data on molecular abnormalities were available for 140 individuals, whereas cytogenetic abnormalities were assessed in a subset of 114 individuals with identified cytogenetic abnormalities. P values are derived from Fisher’s exact test. The association between response (CR/CRi) within 3 cycles and highest variant allele frequency (VAF) per individual (B), number of mutations per individual (C), and number of mutated genes per individual (D). P values are from Wilcoxon signed-rank test.
Associations between molecular and cytogenetic abnormalities (with prevalence ≥10) and response. (A) The association between individual frequently mutated genes (≥10 times) and frequently observed cytogenetic abnormalities (≥10 times) and response (CR/CRi) after 3 cycles. Odds ratios are displayed on a log10 scale, with lines extending to the 95% CI. Horizontal bars indicate the proportion with a mutation in the respective gene for patients with (blue) and without (red) response, with absolute numbers given for each gene. Data on molecular abnormalities were available for 140 individuals, whereas cytogenetic abnormalities were assessed in a subset of 114 individuals with identified cytogenetic abnormalities. P values are derived from Fisher’s exact test. The association between response (CR/CRi) within 3 cycles and highest variant allele frequency (VAF) per individual (B), number of mutations per individual (C), and number of mutated genes per individual (D). P values are from Wilcoxon signed-rank test.
In addition, to address the question of whether the size of the (dominant) clone or the number of (sub) clones affects response to decitabine, we analyzed the relation between variant allele frequency, the number of mutations, and outcome. This analysis revealed that the highest variant allele frequency per individual, the number of mutations per individual (median, 3; range, 0-12), and the number of mutated genes per individual (median, 3; range, 0-9) did not correlate with CR/CRi rates within 3 cycles (Figure 4B-D). With respect to the most common cytogenetic abnormalities, only the presence of complex cytogenetic abnormalities (ie, ≥3) was associated with a greater probability of response (Figure 4A).
Treatment outcome according to minimal residual disease
LAIP was determined by multiparameter flow cytometry in all 144 patients using bone marrow samples before the start of study treatment. Minimal residual disease (MRD) was assessed in bone marrow samples after cycle 3 in 57 patients, of whom 42 were in CR/CRi/MLFS (34 in CR/CRi; 8 in MLFS). Median MRD was 0.12% (range, 0%-47.9%). Of these 57 patients, 28 (49%) became MRD negative (<0.1% LAIP/WBC count). Interestingly, the EFS and OS curves of these 57 patients were independent of their MRD status (positive, n = 29; negative, n = 28) (Figure 5). Also, when only considering the 34 patients in CR/CRi or the 42 patients in CR/CRi/MLFS, the OS was independent of MRD status (supplemental Figure 6). Although the 0.1% threshold was not able to identify patients with superior survival, lowering the cutoff point to 0.05% (resulting in 3 more MRD-positive patients) was not able to discriminate OS between patients with and without MRD.
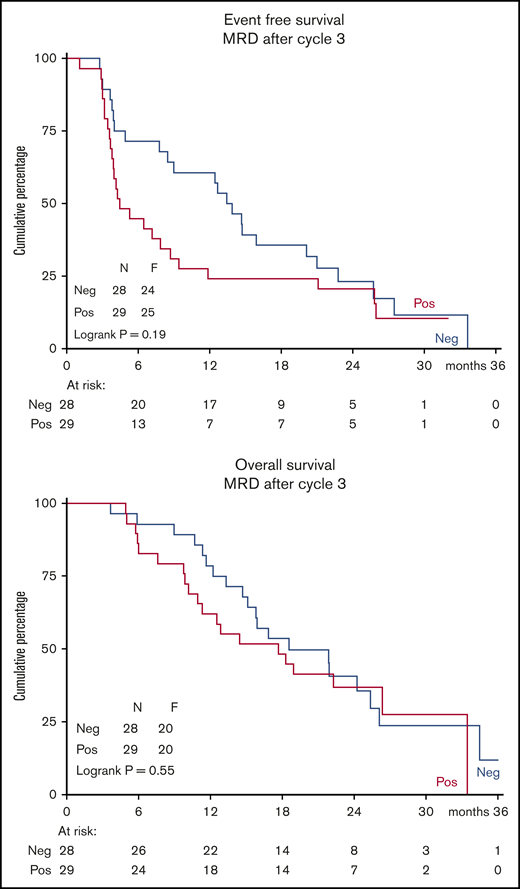
Kaplan-Meier estimates of EFS and OS. Graphs depict data from all 57 patients with available MRD data after 3 cycles.
Kaplan-Meier estimates of EFS and OS. Graphs depict data from all 57 patients with available MRD data after 3 cycles.
Discussion
HMAs are an important treatment modality in older, unfit patients with AML and in higher risk MDS patients. Here we report on the therapeutic value of ibrutinib added to 10-day decitabine regimen. Recent preclinical insights regarding the inhibitory effects of ibrutinib on AML blasts led us to perform a randomized phase 2 clinical trial in older patients with newly diagnosed AML. However, this study, which compared a 10-day decitabine schedule with a combination of decitabine plus ibrutinib, revealed no therapeutic advantage for the ibrutinib combination. No improvement in response was noted for the ibrutinib arm. The results of the study also did not suggest indications for a therapeutic benefit in a particular molecular subset of AML (eg, those with mutated CSF3R or FLT-3). In a previously reported, small, open-label, phase 2a study (n = 36) (accrual between February 2015 and April 2017), ibrutinib alone or in combination with cytarabine or azacitidine had also shown limited antileukemic efficacy.21
Although ibrutinib did not improve the CR/CRi rate (which was the primary end point of the study), this prospective randomized trials shows that 10-day decitabine offers valuable antileukemic activity with a median OS of almost 12 months in this older, unfit patient population. This median OS is comparable with the previously reported median OS that was achieved in so-called fit AML patients of older age with intensive chemotherapy in the HOVON43 study.22 The current outcome data compare favorably with other open-label randomized trials using decitabine, which had a reported median OS of 7.7 months (95% CI, 6.2-9.2) with the 5-day decitabine schedule and 9.3 months (95% CI, 5.8-12.2) with the 10-day schedule.5,23 Although direct comparisons of outcome data across different studies do not allow robust conclusions, the good adherence to the protocol, including start of the second cycle <1 week (independent of peripheral counts) and relatively high number of median cycles of decitabine applied, could have contributed to the comparatively favorable outcome.
As for the study reported here, previous randomized trials failed to show a benefit for additions to HMA with histone deacetylase inhibitors (including entinostat, valproic acid, vorinostat, and pracinostat) and the immune modulating agent lenalidomide and the proteasome inhibitor bortezomib.23-29 Other agents such as selective inhibitors of isocitrate dehydrogenase 1 (ivosidenib) and 2 (enasidenib) in combination with HMAs are currently in advanced clinical testing. The combination of HMA with venetoclax seems to be the first combination that indeed improves outcomes when added to single-agent HMA. The pivotal phase 1b trial evaluating the combination venetoclax plus HMA in untreated AML of the elderly reported acceptable tolerance with high response rates (composite CR rate, 73%).30 In that study, 400 mg of venetoclax seemed to have the best safety profile while providing deep and durable responses in a poor-risk population. The VIALE-A study (#NCT02203773), a randomized phase 3 trial comparing azacitidine plus placebo vs azacitidine plus venetoclax, confirmed the additive value of venetoclax added to azacitidine by a significant increase in the CR/CRi rate from 28% to 66% and a significant increase in median OS from 9.6 months to 14.7 months.31
In our cohort of 144 patients, we were able to identify an association between mutational status and CR/CRi during the first 3 cycles. Of the more frequent mutations (ie, observed ≥10 times), only mutated TP53 was associated with a significantly higher CR/CRi rate, confirming the previously reported favorable response of TP53 mutated AML to HMA.32,33 In contrast to previously published data, in retrospective and smaller cohorts, we could not confirm that mutations in TET2, DNMT3A, and IDH1/2 are associated with higher response rates after treatment with HMA.34-36 Mutated STAG2, IDH2, and ASXL1 were significantly associated with lower CR/CRi rates during the first 3 cycles. In the VIALE-A study, a low CR/CRi rate after treatment with azacitidine was also observed in patients with IDH mutations, which could partially explain the favorable hazard ratio for azacitidine plus venetoclax compared with azacitidine alone for patients with IDH mutations in the VIALE-A study (ie, 0.28 for IDH1 and 0.34 for IDH2-mutated AML).31 Lower CR rates to azacitidine in patients with IDH2 or STAG2 mutations were also observed in a cohort of 250 patients with newly diagnosed, relapsed, or refractory AML or high-risk MDS patients.27 Although the associations with mutated TP53, IDH2, and STAG2 become more solid, in general, the different associations between mutational status and outcome might be explained by the limited numbers, the interaction between the various mutations, and by the heterogeneity of the leukemia within each patient. Finally, although it has been suggested that HMAs are particularly effective against subclones (in contrast to founder clones), it could be hypothesized that the size of the dominant clone or the number of (sub) clones (reflected by the number of mutations) could affect outcome,37,38 although our data did not show this.
In general, responses to HMAs are rarely sustained and have a weak correlation with OS.3-5 Although patients who obtain CR/CRi have a superior OS compared with patients who do not obtain CR/CRi (in our cohort, >12 months longer median OS) (supplemental Figure 2), limited data are available on how the depth of response after HMA correlates with outcome. Our data show that a good quality response, reaching MRD negativity, may be achieved with the 10-day decitabine schedule (49% of the patients after cycle 3). In a cohort of 115 patients with AML treated with various HMAs, of whom 61 patients (53%) had evaluable MRD data at the time of response, 25 (41%) became MRD negative, with no impact on OS.39 In line with these data, in our cohort, MRD negativity did not predict for OS either. This outcome seems discrepant from treatment with intensive chemotherapy in older patients with AML, in whom assessment of treatment response using flow cytometry provides powerful independent prognostic information.40 The clonal dynamics and the predictive value of MRD determined by flow cytometry (and likely NGS) in the context of treatment with intensive chemotherapy and HMA may be different. The relatively mild impact of HMA on founder clones could be an explanation for this difference.37,38
All requests for data sharing should be sent to the corresponding author (Gerwin Huls; e-mail: g.huls@umcg.nl).
Acknowledgment
This investigator-sponsored trial was financially supported by Janssen Pharmaceutics, and they provided the decitabine and ibrutinib used in the trial free of charge.
Authorship
Contribution: The study was designed by the Leukemia Working Group of the HOVON; the HOVON Data Center was responsible for the central data management; D.A.C. and I.v.Z. performed the analysis of the data; G.H., T.P., S.K.K., G.S., L.G., A.A.v.d.L., D.B., D.v.L.-V.., R.B., M.J.-L., M.F., M.H., M.G.M., M.S., R.v.M.K., D.D., M.W.M.v.d.P., M.C.L., L.T., Y.C., E.A., S.B., and G.J.O. collected, analyzed, and interpreted the clinical data; J.C. performed the MRD analyses; P.J.M.V. performed molecular analyses; the decision to publish was made by the cooperative group; and G.H. and subsequently B.L., G.J.O., and D.A.C. produced the first version of the manuscript, which was circulated for comments to the other authors.
Conflict-of-interest disclosure: G.H. has been been involved in advisory boards for Janssen Pharmaceutics. The remaining authors declare no competing financial interests.
A list of the members of the Dutch-Belgian Hemato-Oncology Cooperative Group (HOVON) and Swiss Group for Clinical Cancer Research (SAKK) appears in “Appendix.”
Correspondence: Gerwin Huls, Department of Hematology, University Medical Center Groningen, PO Box 30.001, 9700 RB Groningen, The Netherlands; e-mail: g.huls@umcg.nl.
Appendix: Dutch-Belgian Hemato-Oncology Cooperative Group (HOVON) and Swiss Group for Clinical Cancer Research (SAKK) members
The Netherlands
University Medical Center Groningen, Groningen: G. Huls, I. A. van Zeventer, E. Ammatuna; HOVON Data Center, Erasmus MC Cancer Institute, Rotterdam: D. Chitu; Meander Hospital Amersfoort, Amersfoort: S. K. Klein; Erasmus University Medical Center, Rotterdam: M. Jongen-Lavrencic, B. Löwenberg; Amsterdam University Medical Center, Amsterdam: A. A. van de Loosdrecht, G. Ossenkoppele; Hagaziekenhuis, Den Haag: D. van Lammeren-Venema; Amphia Hospital, Breda: R.S. Boersma; Medical Center Leeuwarden, Leeuwarden: M. Hoogendoorn; Antonius Hospital, Nieuwegein: M. Söhne; Isala Hospital, Zwolle: M. van Marwijk Kooy; Maastricht University Medical Center, Maastricht: M. van der Poel; Medical Spectrum Twente, Enschede: M. C. Legdeur; Maxima Medical Center, Veldhoven: L. Tick; Jeroen Bosch Hospital, Den Bosch: A. Herbers; University Medical Center Utrecht, Utrecht: J. Kuball; Radboud University Medical Center, Nijmegen: W. J. F. M. van der Velden; Rijnstate Hospital, Arnhem: M. Cuijpers; OLVG, Amsterdam: W. E. Terpstra.
Switzerland
nselspital, Bern: T. Pabst; Ospedale Regionale, Billinzona: G. Stussi; Kantonsspital St. Gallen, St. Gallen: M. Fehr; Universitätsspital Zurich, Zurich: M. G. Manz; University Hospital Genève, Genève: Y. Chalandon; University Hospital CHUV, Lausanne: S. Blum; Kantonspital Aarau, Aurau: M. Bargetzi; University Hospital Basel, Basel: D. Heim; Kantonspital Luzern, Luzern: M. Gregor.
Lithuania
Vilnius University Hospital Santaros Klinikos, Vilnius University, Vilnius: L. Griskevicius.
Belgium
ZNA Stuivenberg/Middelheim, Antwerp: D. A. Breems; AZ Delta Roeselare, Roeselare: D. Deeren; Saint Paul Hospital, Haine: B. Bailly.

