

Key Points
NR3C1 KO VSTs are resistant to the lymphocytotoxic effect of glucocorticoids while retaining their phenotype, function, and specificity.
Large-scale production of GMP-grade glucocorticoid-resistant VSTs using CRISPR-Cas9 technology is feasible.

Abstract
Virus-specific T cells have proven highly effective for the treatment of severe and drug-refractory infections after hematopoietic stem cell transplant (HSCT). However, the efficacy of these cells is hindered by the use of glucocorticoids, often given to patients for the management of complications such as graft-versus-host disease. To address this limitation, we have developed a novel strategy for the rapid generation of good manufacturing practice (GMP)–grade glucocorticoid-resistant multivirus-specific T cells (VSTs) using clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) gene-editing technology. We have shown that deleting the nuclear receptor subfamily 3 group C member 1 (NR3C1; the gene encoding for the glucocorticoid receptor) renders VSTs resistant to the lymphocytotoxic effect of glucocorticoids. NR3C1-knockout (KO) VSTs kill their targets and proliferate successfully in the presence of high doses of dexamethasone both in vitro and in vivo. Moreover, we developed a protocol for the rapid generation of GMP-grade NR3C1 KO VSTs with high on-target activity and minimal off-target editing. These genetically engineered VSTs promise to be a novel approach for the treatment of patients with life-threatening viral infections post-HSCT on glucocorticoid therapy.
Introduction
Multivirus-specific T cells (VSTs) have proven safe and effective for the treatment of life-threatening viral infections such as cytomegalovirus (CMV), Epstein-Barr virus, adenovirus, and BK virus (BKV) after allogeneic hematopoietic stem cell transplant (HSCT).1 However, after HSCT, many patients receive steroids for the treatment of complications such as graft-versus-host disease (GVHD). Indeed, after HSCT, viral reactivation often follows the use of glucocorticoids.2 Glucocorticoids are lymphocytotoxic and induce apoptosis of T cells,3 thereby limiting the clinical efficacy of adoptively infused VSTs. Glucocorticoids exert their potent immunosuppressive effect by binding to the glucocorticoid receptor (GR), a pleiotropic ligand-activated transcription factor.4,5 Therefore, engineering VSTs to render them steroid resistant by deleting the nuclear receptor subfamily 3 group C member1 (NR3C1; the gene encoding for the GR protein) could be a valid strategy for the treatment of patients with life-threatening viral infections receiving glucocorticoid therapy.
Here, we present a novel strategy for the production of good manufacturing practice (GMP)-grade VSTs that have been engineered to silence the expression of the GR using clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated protein 9 (Cas9) gene editing.
Methods
VST generation
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll from buffy coats of seropositive donors. PBMCs were then stimulated without selection with virus-specific PepMix (1 μg/mL; JPT Peptide Technologies) and cultured in RPMI 1640 plus 10% human AB serum at 37°C with 5% CO2 for 2 hours. Two hours later, a cytokine cocktail was added (10 ng/mL interleukin 7 [IL-7], 50 IU/mL IL-2, 10 ng/mL IL-15). Media were changed (RPMI 1640 plus 10% human AB serum) and fresh cytokines were added every 2 days for the length of culture.
CRISPR-Cas9 gene editing: small scale
NR3C1 knockout (KO) was performed on days 7 to 10 of VST expansion using the ribonucleoprotein (RNP) complex. We used 2 CRISPR RNAs (crRNAs) targeting exon 2 of the human GR gene: crRNA#1, TGAGAAGCGACAGCCAGTGA; crRNA#2, GGCCAGACTGGCACCAACGG. First, the crRNA plus trans-activating crRNA (tracrRNA) duplex for each crRNA was prepared by incubation at 95°C for 5 minutes in a thermocycler at equimolar concentrations. Cas9 protein (Integrated DNA Technologies [IDT]) and guide RNA (gRNA; crRNA plus tracrRNA combination) were incubated at room temperature for 15 minutes in a 1:1 ratio.6 The incubation product (gRNA and Cas9 complex) was then used to electroporate 1 million to 2 million VSTs using the Neon Transfection System (Thermo Fisher Scientific). Alt-R HiFi Cas9 Nuclease 3-NLS (IDT) was also used in this protocol to form RNP complexes with gRNAs for electroporation into VSTs. Optimized electroporation conditions were 1600 V, 10 milliseconds, 3 pulses, using T buffer. For the off-target studies of gRNA#1 and gRNA#2, Alt-R crRNA and ATTO-labeled tracrRNA (IDT) comprised the gRNA; RNP complexes were formed with both Alt-R wild-type (WT) and Alt-R HiFi Streptococcus pyogenes (S.p.) Cas9 and electroporated into VSTs.
CRISPR-Cas9 gene editing: GMP-compliant, large scale
For the large-scale CRISPR-Cas9 protocol, we used the 4D Nucleofector from Lonza. The program that was adopted was EO-115 and the buffer used was P3 buffer for primary cells. As in the small-scale CRISPR-Cas9 protocol, we first prepared the crRNA plus tracrRNA duplex for each crRNA by incubating them at 95°C for 5 minutes in a thermocycler at equimolar concentrations. Cas9 protein (IDT) and gRNA (crRNA plus tracrRNA combination) were then incubated at room temperature for 15 minutes. To electroporate 5 × 106 cells, we used 2.2 μM Cas9 and 2.4 μM gRNA. For the larger cell doses, reagents were then scaled up by dividing the cell dose of interest by 5 × 106 and then multiplying this number by the final amount of RNP complex used to electroporate 5 × 106 cells.
PCR gel electrophoresis
DNA was extracted and purified (QIAamp DNA Blood Mini Kit; Qiagen Inc) from VSTs (control and NR3C1 KO conditions). We used the Platinum SuperFi Green PCR Master Mix from Invitrogen for polymerase chain reaction (PCR) amplification using the following PCR primers spanning the Cas9–single-guide RNA cleavage site of exon 2 of the GR gene: exon 2 forward primer, GGACTCCAAAGAATCATTAACTCCTGG; exon 2 reverse primer, AATTACCCCAGGGGTGCAGA.
DNA bands were separated by polyacrylamide gel electrophoresis prepared with SYBR-safe DNA gel stain in 0.5× Tris/Borate/EDTA. Gel images were obtained using a GBox machine with GeneSys software (Syngene). Band intensities were analyzed using Image J software by plotting the band intensities for each lane. The KO efficiency for NR3C1 (percentage) was calculated by dividing the intensity of the cleaved band by the intensity of the uncleaved control band.
Western blot
To detect GR protein expression, VSTs were lysed in lysis buffer (IP Lysis Buffer; Pierce Biotechnology Inc) supplemented with protease inhibitors (Complete Mini, EDTA-free Cocktail tablets; Roche Holding) and incubated for 30 minutes on ice. Protein concentrations were determined by the bicinchoninic acid assay (Pierce Biotechnology Inc). The following primary antibodies were used: GR (clone D6H2L) XP rabbit monoclonal antibody and β-actin antibody (clone 8H10D10); both antibodies were obtained from Cell Signaling Technology. Blots were imaged using the LI-COR Odyssey Infrared Imaging System. Bands were quantified using Image J software. The percentage (%) of GR protein loss was calculated relative to values in control cells and normalized to β-actin loading control.
Flow cytometry
The following antibodies were used for flow cytometry staining: anti-human CD3 antibody (BV650, clone UCHT1; BD Biosciences), anti-human CD4 antibody (allophycocyanin [APC], clone RPA-T4; Invitrogen), anti-human CD8 antibody (peridinin chlorophyll protein [Percp], clone SK1; BioLegend), anti-human CD62L (BV605, clone DREG-56; BD Biosciences), anti-human CD45RA (phycoerythrin-Cy7, clone HI100; BD Biosciences), and anti-human CCR7 (fluorescein isothiocyanate, clone G03H7; BioLegend). All data were acquired with BD Fortessa (BD Biosciences) and analyzed with FlowJo software.
Annexin V apoptosis assay
To evaluate the effect of dexamethasone on the viability of VSTs (Cas9 control or NR3C1 KO), the annexin V apoptosis assay was performed. Cas9 control or NR3C1 KO VSTs were treated with dexamethasone (200 μM; Sigma) for 72 hours, the cells were collected and washed with annexin V buffer and stained with annexin V (V500; BD Biosciences) and live/dead (efluor 660; Invitrogen) in addition to CD3 (BV650, clone UCHT1; BD Biosciences), CD4 (APC, RPA-T4; Invitrogen), and CD8 (Percp, clone SK1; BioLegend). After gating on T cells, the proportion of apoptotic (positive for annexin V) and dead cells (positive for live/dead stain) was determined by flow cytometry.
Functional assays
VSTs were seeded at 100 × 103 cells per 200 μL per well in the presence of brefeldin A for 6 hours in round-bottom 96-well plates with viral PepMix. Cells were then collected and washed with phosphate-buffered saline and surface and intracellular staining was performed using a BD fixation/permeabilization kit. Cells were stained with live/dead aqua dead cell stain (Thermo Fisher Scientific), anti-human CD3 antibody (BV650, clone UCHT1; BD Biosciences), anti-human CD4 antibody (APC, clone RPA-T4; Invitrogen), and anti-human CD8 antibody (Percp, clone SK1; BioLegend). CD107a degranulation was measured with anti-human CD107a (LAMP-1) conjugated to Brilliant Violet 785 (BioLegend), which was added to the wells at the beginning of the assay. Intracellular cytokine production was determined using antibodies against tumor necrosis factor-α (TNF-α; clone MAb11 conjugated to Alexa Fluor 700; eBioscience Inc), interferon-γ (IFN-γ; conjugated to V450, clone B27; BD Biosciences), and IL-2 (conjugated to phycoerythrin, clone MQ1-17H12; BD Biosciences) by flow cytometry as previously described.7
In vivo xenograft model
To test the engraftment and persistence of NR3C1 KO VSTs in vivo, 4 groups of NSG mice (10-week-old female, n = 5 per group) were irradiated on day −1 and then injected on day 0 with either 1 × 106 control VSTs (n = 10, 2 groups) or 1 × 106NR3C1 KO VSTs (n = 10, 2 groups). One-half of the mice (1 group per condition) were then monitored and the other half were treated with 15 mg/kg daily dexamethasone subcutaneously for 2 weeks. The mice were then euthanized and the bone marrow harvested for flow cytometry staining. The antibodies used for the in vivo studies were the following: live dead aqua (Thermo Fisher Scientific), anti-human CD45 antibody (conjugated to Percp, clone HI30; BioLegend), anti-human CD3 antibody (BV650, clone UCHT1; BD Biosciences), anti-human CD4 antibody (APC, clone RPA-T4; Invitrogen), and anti-human CD8 antibody (Percp, clone SK1; BioLegend).
Off-target identification
The GUIDE-seq method was used for unbiased discovery of off-target editing events.8 In this study, HEK293 cells that constitutively express the S.p. Cas9 nuclease (“HEK293-Cas9” cells) were used as the source of Cas9. Alt-R gRNA complexes were formed by combining Alt-R tracrRNA and Alt-R crRNA XT at a 1:1 molar ratio. gRNA complexes were delivered by nucleofection using the Amaxa Nucleofector 96-well Shuttle System (Lonza). For each nucleofection, 3.5 × 105 HEK293-Cas9 cells were washed with 1× phosphate-buffered saline, resuspended in 20 µL of solution SF (Lonza), and combined with 10 µM gRNA together with 0.5 µM GUIDE-seq double-stranded DNA donor fragment. This mixture was transferred into 1 well of a Nucleocuvette plate (Lonza) and electroporated using protocol 96-DS-150. DNA was extracted 72 hours after electroporation using the GeneJET Genomic DNA purification kit (Thermo Fisher Scientific). Next-generation sequencing (NGS) library preparation, sequencing, and operation of the GUIDE-seq software was performed as previously described,9 with the additional inclusion of the Needleman-Wunch alignment.
Target enrichment via rhAmpSeq for multiplexed PCR
To quantify editing at off-target sites identified with GUIDE-seq, we performed multiplex PCR coupled to amplicon NGS, using PCR executed in the presence of RNase H210 with blocked-cleavable primers. Primers were designed by an algorithm (developed by IDT) for primer cross-comparison and selection based on compatibility with other primers in the multiplex. This amplification technology requires that the primer properly hybridizes to a target site before amplification. Mismatches between target and primer prevent unblocking, thereby increasing specificity and eliminating primer dimers. This approach enables efficient production of highly multiplex PCR amplicons in a single tube. For these experiments, gRNA complexes were delivered into HEK293-Cas9 cells as previously described or complexed with Alt-R HiFi Cas9 nuclease v3 to form an active RNP complex, which was then directly nucleofected into HEK293 cells at 2 µM together with 2 µM Alt-R Cas9 Electroporation Enhancer (IDT). DNA was extracted 48 hours after electroporation using QuickExtract DNA Extraction Solution (Epicentre). Genomic DNA was extracted in the same way from natural killer cells for off-target studies comparing WT and HiFi Alt-R Cas9 RNP complexes with editing mediated by optimized delivery conditions, as described previously.11 Locus-specific amplification with RNase H2 primers was performed for 10 cycles followed by a 1.3× solid-phase reversible immobilization bead clean-up. An indexing round of PCR was performed for 18 cycles to incorporate sample-unique P5 and P7 indexes followed by a 1× solid-phase reversible immobilization bead clean-up and library quantification by quantitative PCR (IDT). PCR amplicons were sequenced on an Illumina MiSeq instrument (v2 chemistry, 150-bp paired-end reads) (Illumina). Data were analyzed using a custom-built pipeline. Data were demultiplexed (Picard tools v2.9; https://github.com/broadinstitute/picard); forward and reverse reads were merged into extended amplicons (flash v1.2.11). Reads were aligned against the GRCh38 genomic reference (minimap2 v2.12), and were assigned to targets in the multiplex primer pool (bedtools tags v2.25). Reads were realigned to the target, favoring alignment choices with insertions-deletions near the Cas9-predicted cut site. At each target, editing was calculated as the percentage of total reads containing an insertion-deletion within a 4-bp window of the cut site.
Statistics
The 2-way analysis of variance test or the Student t test was used as appropriate to compare quantitative differences (mean plus or minus standard deviation [SD]) between groups; P values were 2-sided and P < .05 was considered significant. The indicated statistical tests were performed using Prism software (GraphPad version 7.0c). For the confocal microscopy analysis, data sets were analyzed using the unpaired Student t test. Data present mean plus or minus the 95% confidence interval. Images were assembled using ImageJ.
Results
Generation of NR3C1 KO VSTs using CRISPR-Cas9 technology
To generate NR3C1 KO VSTs against CMV, BKV, and adenovirus, PBMCs were isolated from seropositive donors and cultured in vitro with PepMix from the immunodominant proteins of the 3 viruses in the presence of a cytokine cocktail (50 IU/mL IL-2, 10 ng/mL IL-7, 10 ng/mL IL-15). On days 7 to 10 of cell expansion, we used RNP-mediated CRISPR-Cas9 gene editing to silence NR3C1 in T cells (Figure 1A-B). Two crRNA sequences were used to target exon 2 of the human NR3C1 gene located on chromosome 5 (Figure 1B). The efficiency of NR3C1 KO was high (82% to 98%) as determined by PCR (Figure 1C) and western blot analysis (Figure 1D).
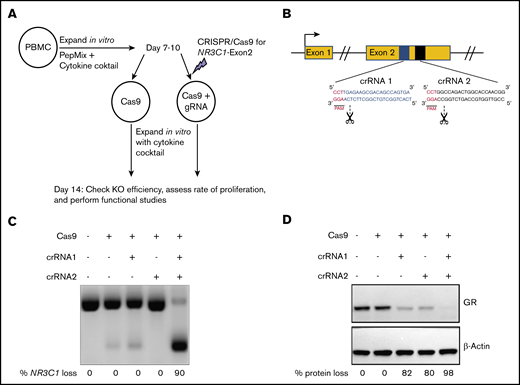
CRISPR-Cas9–mediated deletion of the gene coding for the GR in primary human VSTs. (A) Schematic summary of the protocol for CRISPR-Cas9–mediated KO of NR3C1 in primary human VSTs. PBMCs were stimulated with virus-specific PepMix from CMV, BKV, and adenovirus (in combination) in the presence of 10 ng/mL IL-7, 50 IU/mL IL-2, and 10 ng/mL IL-15. On days 7 to 10 of in vitro expansion, primary human VSTs were nucleofected with control Cas9 alone (Cas9 control) or Cas9 preloaded with gRNA targeting the exon 2 of the NR3C1 gene, which encodes for the GR protein. KO efficiency and functional assays were performed at day +14 post initial PBMC isolation. (B) Schematic representation of CRISPR-Cas9–mediated NR3C1 KO using 2 short guide crRNAs targeting the exon 2 of NR3C1 gene. (C-D) The NR3C1 KO efficiency of VSTs after electroporation with Cas9 alone (control), Cas9 complexed with 1 crRNA (crRNA 1 or crRNA 2), or Cas9 complexed with the combination of 2 crRNAs (crRNA 1 plus crRNA 2) using a WT Cas9 was determined by PCR analysis at day 3 (C) or western blot analysis at day 7 (D) after electroporation. The percentages (%) of NR3C1 KO efficiency (C) and GR protein loss (D) after CRISPR-Cas9 gene editing are shown under each figure. PAM, protospacer adjacent motif.
CRISPR-Cas9–mediated deletion of the gene coding for the GR in primary human VSTs. (A) Schematic summary of the protocol for CRISPR-Cas9–mediated KO of NR3C1 in primary human VSTs. PBMCs were stimulated with virus-specific PepMix from CMV, BKV, and adenovirus (in combination) in the presence of 10 ng/mL IL-7, 50 IU/mL IL-2, and 10 ng/mL IL-15. On days 7 to 10 of in vitro expansion, primary human VSTs were nucleofected with control Cas9 alone (Cas9 control) or Cas9 preloaded with gRNA targeting the exon 2 of the NR3C1 gene, which encodes for the GR protein. KO efficiency and functional assays were performed at day +14 post initial PBMC isolation. (B) Schematic representation of CRISPR-Cas9–mediated NR3C1 KO using 2 short guide crRNAs targeting the exon 2 of NR3C1 gene. (C-D) The NR3C1 KO efficiency of VSTs after electroporation with Cas9 alone (control), Cas9 complexed with 1 crRNA (crRNA 1 or crRNA 2), or Cas9 complexed with the combination of 2 crRNAs (crRNA 1 plus crRNA 2) using a WT Cas9 was determined by PCR analysis at day 3 (C) or western blot analysis at day 7 (D) after electroporation. The percentages (%) of NR3C1 KO efficiency (C) and GR protein loss (D) after CRISPR-Cas9 gene editing are shown under each figure. PAM, protospacer adjacent motif.
Silencing GR protects VSTs from the lymphocytotoxic effect of glucocorticoids without altering their phenotype or function
We next determined whether silencing the GR protects VSTs from the lymphocytotoxic effect of glucocorticoids. NR3C1 KO VSTs or control VSTs (defined in this work as VSTs electroporated with Cas9 alone) were cultured with 200 μM dexamethasone for 72 hours and their viability was assessed using the annexin V apoptosis assay. At the end of culture, the majority of control VSTs were either apoptotic or dead, whereas NR3C1 KO cells remained viable, confirming that NR3C1 KO VSTs are resistant to the lymphocytotoxic effect of glucocorticoids (Figure 2A-C).
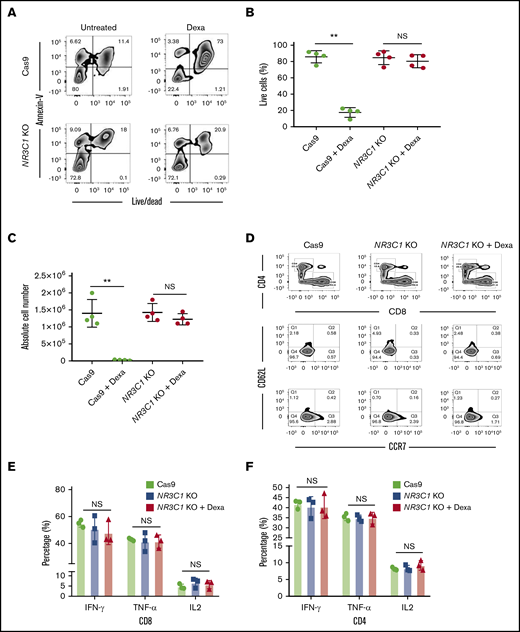
NR3C1 deletion renders VSTs resistant to steroids without altering their phenotype or function. (A) Representative fluorescence-activated cell sorter (FACS) plots showing the percentage of apoptotic cells (annexin V+) and alive or dead cells (live/dead stain) in control Cas9 vs NR3C1 KO (exon 2) VSTs after culture with or without dexamethasone (Dexa; 200 μM) for 72 hours (n = 4). Inset values indicate the percentage of annexin V and alive/dead cells from each group. (B-C) Graph summarizing the percentage of live cells (B) and the absolute cell number (C) between control Cas9 and NR3C1 KO VSTs treated with or without 200 μM dexamethasone for 72 hours (n = 4). Statistical significance is indicated as **P ≤ .01. Bars represent mean values with standard deviation. (D) FACS plots showing the distribution of CD4 and CD8 (upper plots) and the frequency of CD62L and CCR7 (lower plots) in control Cas9 or NR3C1 KO VSTs that were treated with or without Dexa (200 μM) for 72 hours (n = 3). Inset values indicate the percentage of T cells expressing CD62L and/or CCR7 in each treatment group. These FACS plots were pregated on CD3+CD45RA− T cells. (E-F) Bar graphs showing the percentage of IFN-γ, TNF-α, or IL-2 production by CD8+ (E) and CD4+ (F) VSTs treated with control Cas9 (green), NR3C1 KO (blue), or NR3C1 KO plus dexamethasone (Dexa; 200μM; red) in response to 6 hours of stimulation with viral PepMix (n = 3). The functional analysis of the Cas9+Dexa group was not performed due to the absence of viable cells resulting from the lymphocytotoxic effect of steroids. The bars represent mean values with SD. NS, not significant.
NR3C1 deletion renders VSTs resistant to steroids without altering their phenotype or function. (A) Representative fluorescence-activated cell sorter (FACS) plots showing the percentage of apoptotic cells (annexin V+) and alive or dead cells (live/dead stain) in control Cas9 vs NR3C1 KO (exon 2) VSTs after culture with or without dexamethasone (Dexa; 200 μM) for 72 hours (n = 4). Inset values indicate the percentage of annexin V and alive/dead cells from each group. (B-C) Graph summarizing the percentage of live cells (B) and the absolute cell number (C) between control Cas9 and NR3C1 KO VSTs treated with or without 200 μM dexamethasone for 72 hours (n = 4). Statistical significance is indicated as **P ≤ .01. Bars represent mean values with standard deviation. (D) FACS plots showing the distribution of CD4 and CD8 (upper plots) and the frequency of CD62L and CCR7 (lower plots) in control Cas9 or NR3C1 KO VSTs that were treated with or without Dexa (200 μM) for 72 hours (n = 3). Inset values indicate the percentage of T cells expressing CD62L and/or CCR7 in each treatment group. These FACS plots were pregated on CD3+CD45RA− T cells. (E-F) Bar graphs showing the percentage of IFN-γ, TNF-α, or IL-2 production by CD8+ (E) and CD4+ (F) VSTs treated with control Cas9 (green), NR3C1 KO (blue), or NR3C1 KO plus dexamethasone (Dexa; 200μM; red) in response to 6 hours of stimulation with viral PepMix (n = 3). The functional analysis of the Cas9+Dexa group was not performed due to the absence of viable cells resulting from the lymphocytotoxic effect of steroids. The bars represent mean values with SD. NS, not significant.
Next, we examined whether silencing the GR affects the phenotype or function of the CD4+ and CD8+ VSTs. Flow cytometry analysis confirmed that compared with control VSTs, NR3C1 KO did not affect the distribution of CD4+ and CD8+ or the maturational profile of VSTs (Figure 2D). NR3C1 KO VSTs had a comparable effector function to control VSTs as measured by production of IFN-γ, TNF-α, or IL-2 in response to ex vivo stimulation with viral antigens (Figure 2E-F; supplemental Figure 1A-B). Furthermore, culture of NR3C1 KO VSTs in the presence of dexamethasone did not affect their effector function against the relevant viral antigens (Figure 2E-F). These findings confirm that CRISPR-Cas9–mediated knockout of NR3C1 in VSTs protects them from steroid-induced lymphocytotoxicity while preserving their phenotype, function, and specificity.
CRISPR-Cas9–modified NR3C1 KO VSTs are resistant to glucocorticoids in an in vivo xenograft model
We next tested the in vivo persistence of NR3C1 KO VSTs and their resistance to systemic dexamethasone therapy using an immunodeficient mouse model. NSG mice were sublethally irradiated on day −1; on day 0, they received either 1 × 106 control VSTs electroporated with Cas9 alone (n = 10) or 1 × 106NR3C1 KO VSTs (n = 10). Five mice in each group were then treated with 15 mg/kg daily dexamethasone subcutaneously for 2 weeks, and the remaining mice in each group were observed (Figure 3A). At the end of treatment, the animals were euthanized and their bone marrow harvested for T-cell enumeration by flow cytometry. In the animals that received control VSTs, as expected, human CD3+ T cells could only be detected in the group that did not receive dexamethasone (Figure 3B-C). In contrast, in the animals that received NR3C1 KO VSTs, human T cells were present at high frequencies (and in comparable numbers) in all animals, irrespective of whether they were treated with dexamethasone or not (Figure 3B-C). Furthermore, human T-cell frequencies in mice that received the NR3C1 KO VSTs were similar to those seen in animals treated with unmodified VSTs in the absence of dexamethasone (Figure 3B-C). We did not observe any evidence of toxicity including GVHD on necropsies in any of the experimental groups (supplemental Figure 2A-F).
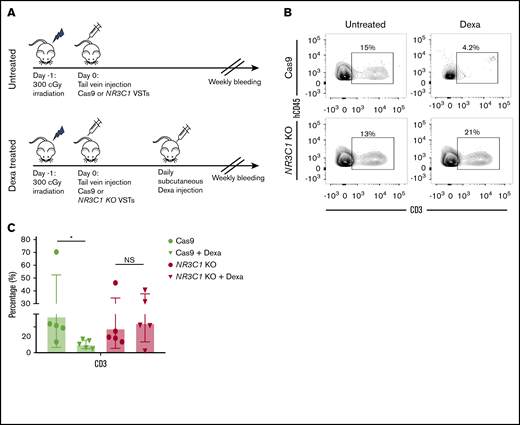
NR3C1 KO VSTs persist in vivo even after treatment with dexamethasone. (A) Schematic diagram representing the timeline for the in vivo experiments. NSG mice received 1 dose (1 × 106) of control Cas9 or NR3C1 KO VSTs and were either observed (top panel) or treated with a daily subcutaneous dose of dexamethasone (15 mg/kg) for 2 weeks (bottom panel) (n = 5 mice per group). (B) All mice were euthanized on day +14 after the adoptive infusion of Cas9 control or NR3C1 KO VSTs. Cells were collected from the bone marrow and analyzed by flow cytometry for the expression of human CD45 (hCD45) and CD3. Representative FACS plots are presented. Inset values indicate the percentage of CD3+ cells from each group. (C) Bar graph shows the pooled data for the percentage of CD3+ cells from panel B (n = 5). The bars represent mean values with SD. The statistical significance is indicated as *P ≤ .05.
NR3C1 KO VSTs persist in vivo even after treatment with dexamethasone. (A) Schematic diagram representing the timeline for the in vivo experiments. NSG mice received 1 dose (1 × 106) of control Cas9 or NR3C1 KO VSTs and were either observed (top panel) or treated with a daily subcutaneous dose of dexamethasone (15 mg/kg) for 2 weeks (bottom panel) (n = 5 mice per group). (B) All mice were euthanized on day +14 after the adoptive infusion of Cas9 control or NR3C1 KO VSTs. Cells were collected from the bone marrow and analyzed by flow cytometry for the expression of human CD45 (hCD45) and CD3. Representative FACS plots are presented. Inset values indicate the percentage of CD3+ cells from each group. (C) Bar graph shows the pooled data for the percentage of CD3+ cells from panel B (n = 5). The bars represent mean values with SD. The statistical significance is indicated as *P ≤ .05.
Analysis of CRISPR-Cas9 off-target cleavage events in VSTs
Off-target genome editing is a potential hurdle for the translation of the CRISPR gene-editing approach to the clinic. To evaluate the landscape of genome-wide off-target effects for the specific NR3C1 crRNAs used in this study, we performed experimental off-target validation using GUIDE-seq and rhAmpSeq technologies (IDT). GUIDE-seq experiments were performed using HEK293 cells that constitutively express S.p. Cas9 nuclease paired with highly modified synthetic gRNAs. The previously published methods8 were otherwise followed to identify the off-target sites associated with each crRNA that had the highest potential to be edited (Figure 4A). Using the rhAmpSeq system, a multiplexed targeted amplicon-sequencing technology, we then focused on the sites identified by GUIDE-seq to more comprehensively investigate the off-target gene-editing events in VSTs electroporated with RNP complexes targeting the NR3C1 locus. Cells treated with WT S.p. Cas9 protein had a low frequency of off-target editing events with either crRNA1, crRNA2 (Figure 4B), or the combination of both crRNAs (Figure 4C). The use of a high-fidelity Cas9 (Alt-R HiFi Cas9 v3; IDT) protein, a Cas9 variant that displays superior on- to off-target ratio when delivered in RNP format,11 resulted in efficient KO (supplemental Figure 3A-B) and further reduced the incidence of off-target events to <0.5% (Figure 4B-C). The high on-target editing activity associated with minimal off-target events supports the translation of this approach to the clinic.
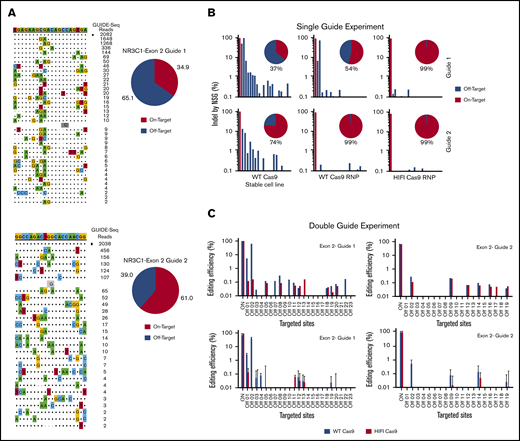
Identification of Cas9 off-target sites by GUIDE-seq and quantification of potential Cas9 off-target cleavage sites using rhAmpSeq technology. (A) Sequences of off-target sites identified by GUIDE-seq for 2 gRNAs targeting the NR3C1 locus (gRNA#1, top panel; gRNA#2, bottom panel). The guide sequence is listed on top with off-target sites shown below. The on-target site is identified with a black square. Mismatches to the guide are shown and highlighted in color with insertions shown in gray below. The number of GUIDE-seq sequencing reads are shown to the right of each site. Pie charts indicate the fractional percentage of the total unique, CRISPR-Cas9–specific read counts that are on-target (orange) and off-target (blue). (B) On- and off-target effects were determined by targeted amplification followed by NGS for exon 2–guide 1 and for exon 2–guide 2. Individual gRNAs were delivered into HEK293 cells stably expressing WT-Cas9 (left panels), or delivered into HEK293 cells by complexing to WT-Cas9 protein (middle panels) or HiFi-Cas9 protein (right panels). Pie charts indicate the percentage of on-target effect (in red) and off-target effect (in blue). (C) Editing efficiency was determined using targeted amplification followed by NGS. Exon 2–guide 1 and exon 2–guide 2 gRNAs were complexed simultaneously with either WT-Cas9 (blue bars) or HiFi-Cas9 (red bars) into HEK293 cells. Editing efficiencies were determined for the known on- and off-target sites for exon 2–guide 1 (left top panel), and exon 2–guide 2 (right top panel). In a similar experiment, exon 2–guide 1 and exon 2–guide 2 gRNAs were complexed simultaneously with either WT-Cas9 (blue bars) or HiFi-Cas9 (red bars) into primary human T cells. Editing efficiencies were determined for the known on- and off-target sites for exon 2–guide 1 (left bottom panel), and exon 2–guide 2 (right bottom panel). Data are shown as mean and SD from n = 3 human donors.
Identification of Cas9 off-target sites by GUIDE-seq and quantification of potential Cas9 off-target cleavage sites using rhAmpSeq technology. (A) Sequences of off-target sites identified by GUIDE-seq for 2 gRNAs targeting the NR3C1 locus (gRNA#1, top panel; gRNA#2, bottom panel). The guide sequence is listed on top with off-target sites shown below. The on-target site is identified with a black square. Mismatches to the guide are shown and highlighted in color with insertions shown in gray below. The number of GUIDE-seq sequencing reads are shown to the right of each site. Pie charts indicate the fractional percentage of the total unique, CRISPR-Cas9–specific read counts that are on-target (orange) and off-target (blue). (B) On- and off-target effects were determined by targeted amplification followed by NGS for exon 2–guide 1 and for exon 2–guide 2. Individual gRNAs were delivered into HEK293 cells stably expressing WT-Cas9 (left panels), or delivered into HEK293 cells by complexing to WT-Cas9 protein (middle panels) or HiFi-Cas9 protein (right panels). Pie charts indicate the percentage of on-target effect (in red) and off-target effect (in blue). (C) Editing efficiency was determined using targeted amplification followed by NGS. Exon 2–guide 1 and exon 2–guide 2 gRNAs were complexed simultaneously with either WT-Cas9 (blue bars) or HiFi-Cas9 (red bars) into HEK293 cells. Editing efficiencies were determined for the known on- and off-target sites for exon 2–guide 1 (left top panel), and exon 2–guide 2 (right top panel). In a similar experiment, exon 2–guide 1 and exon 2–guide 2 gRNAs were complexed simultaneously with either WT-Cas9 (blue bars) or HiFi-Cas9 (red bars) into primary human T cells. Editing efficiencies were determined for the known on- and off-target sites for exon 2–guide 1 (left bottom panel), and exon 2–guide 2 (right bottom panel). Data are shown as mean and SD from n = 3 human donors.
Clinical-scale manufacture of GMP-grade NR3C1 KO VSTs
Next, we developed a strategy to produce clinically relevant numbers of GMP-compliant NR3C1 KO VSTs. The protocol was optimized using GMP-compliant material, including clinical-grade gRNA (Synthego) and SpyFi Cas9 (the GMP version of HiFi Cas9; Aldevron). The scale-up protocol is detailed in “Methods.” Briefly, we expanded the T cells using good laboratory practice (GLP)–grade PepMix from the immunodominant proteins of CMV, BKV, and adenovirus. On days 7 to 10 of expansion, different VST numbers (3 × 106, 25 × 106, and 100 × 106) were electroporated in the presence of Cas9 and gRNA using the Lonza 4D nucleofector. The cells were expanded for another 4 to 7 days and then harvested for functional studies. The KO efficiency was high (93% to 99%) and equivalent at both the DNA and protein levels at the 3 dose levels tested (Figure 5A-B). Functional studies revealed that, at all dose levels tested, the viability and proliferation capacity of GMP-grade NR3C1 KO VSTs, even when cultured with dexamethasone, was similar to that of controls (Figure 5C-D). Furthermore, the NR3C1 KO VSTs generated with large-scale nucleofection maintained similar distribution of CD4 and CD8 compartments compared with control VSTs, even after treatment with dexamethasone (supplemental Figure 4). Moreover, large-scale NR3C1 KO VSTs preserved their effector function and their ability to produce IFN-γ, TNF-α, and IL-2 in response to stimulation with the virus PepMix in both the CD8 and CD4 T-cell compartments (Figure 5E-F). These data prove that scale-up of this strategy using GMP-grade materials is technically feasible and attractive for clinical translation. The ultimate goal is to generate a large biobank of next-generation GMP grade NR3C1 KO VSTs to treat viral infections in immunosuppressed patients receiving glucocorticoid therapy (see visual abstract).
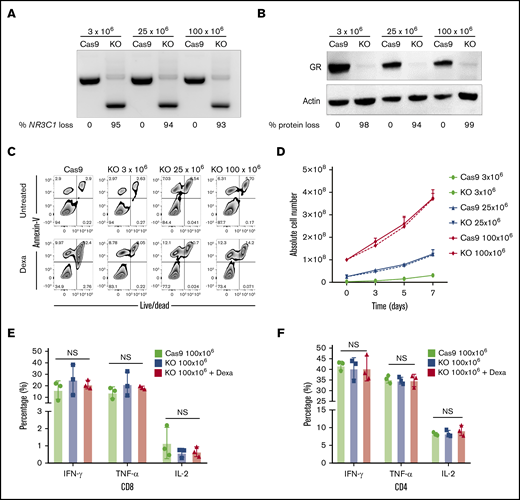
Successful scale-up of CRISPR-Cas9–mediated NR3C1 deletion in VSTs. (A) Different VST numbers (3 × 106, 25 × 106, and 100 × 106) were electroporated in the presence of Cas9 and gRNA using the Lonza 4D nucleofector and the NR3C1 KO efficiency in Cas9 (control) or NR3C1 KO VSTs was determined using PCR (A; n = 3) and western blot analysis (B; n = 3). β-actin was used as loading control in panel B. The percentages (%) of NR3C1 KO efficiency (A) and GR protein loss (B) after CRISPR-Cas9 gene editing are shown under each figure. (C) Representative FACS plots showing the percentage of apoptotic cells (annexin V+) and alive or dead cells (live/dead stain) in control Cas9 and NR3C1 KO VST cells at the different cell dose levels of 3 × 106, 25 × 106, or 100 × 106 cells per electroporation treated with or without Dexa (200 μM) for 72 hours (n = 3). (D) Graph summarizing the absolute cell number between control Cas9 (solid lines) and NR3C1 KO VST cells (dotted lines) at the different cell dose levels of 3 × 106 (green), 25 × 106 (blue), or 100 × 106 (red) cells per electroporation for 72 hours (n = 3). Bars represent mean values with SD. (E-F) Bar graphs showing the percentage of IFN-γ, TNF-α, or IL-2 production by 100 × 106 control Cas9 (green), 100 × 106NR3C1 KO (blue), or 100 × 106NR3C1 KO plus dexamethasone (Dexa 200 μM; red) VSTs in response to 6-hour stimulation with the relevant viral PepMix in the CD8+ T-cell (E) and CD4+ T-cell (F) compartments (n = 3). The functional analysis of the Cas9+Dexa group was not performed due to the absence of viable cells resulting from the lymphocytotoxic effect of steroids. The bars represent mean values with SD.
Successful scale-up of CRISPR-Cas9–mediated NR3C1 deletion in VSTs. (A) Different VST numbers (3 × 106, 25 × 106, and 100 × 106) were electroporated in the presence of Cas9 and gRNA using the Lonza 4D nucleofector and the NR3C1 KO efficiency in Cas9 (control) or NR3C1 KO VSTs was determined using PCR (A; n = 3) and western blot analysis (B; n = 3). β-actin was used as loading control in panel B. The percentages (%) of NR3C1 KO efficiency (A) and GR protein loss (B) after CRISPR-Cas9 gene editing are shown under each figure. (C) Representative FACS plots showing the percentage of apoptotic cells (annexin V+) and alive or dead cells (live/dead stain) in control Cas9 and NR3C1 KO VST cells at the different cell dose levels of 3 × 106, 25 × 106, or 100 × 106 cells per electroporation treated with or without Dexa (200 μM) for 72 hours (n = 3). (D) Graph summarizing the absolute cell number between control Cas9 (solid lines) and NR3C1 KO VST cells (dotted lines) at the different cell dose levels of 3 × 106 (green), 25 × 106 (blue), or 100 × 106 (red) cells per electroporation for 72 hours (n = 3). Bars represent mean values with SD. (E-F) Bar graphs showing the percentage of IFN-γ, TNF-α, or IL-2 production by 100 × 106 control Cas9 (green), 100 × 106NR3C1 KO (blue), or 100 × 106NR3C1 KO plus dexamethasone (Dexa 200 μM; red) VSTs in response to 6-hour stimulation with the relevant viral PepMix in the CD8+ T-cell (E) and CD4+ T-cell (F) compartments (n = 3). The functional analysis of the Cas9+Dexa group was not performed due to the absence of viable cells resulting from the lymphocytotoxic effect of steroids. The bars represent mean values with SD.
Discussion
Viral reactivation is a common complication after HSCT with significant risks of morbidity and mortality.12 Conventional antiviral treatments are either toxic (eg, ganciclovir or foscarnet for CMV) or have limited efficacy (eg, for adenovirus or BKV). Thus, a number of centers have developed VSTs and have shown the feasibility, safety, and efficacy of this approach to treat viral infections post-HSCT.1,13,14 Viral reactivation is often triggered by the administration of glucocorticoids for the management of complications post-HSCT such as GVHD,2,15 making such patients ineligible for VST therapy. Herein, we have developed a novel strategy for the generation of glucocorticoid-resistant VSTs by deleting the NR3C1 gene using CRISPR-Cas9 RNP-mediated gene-editing technology. This approach was highly efficient with negligible off-target events. We have shown that NR3C1 KO VSTs are resistant to the lymphocytotoxic effect of glucocorticoids both in vitro and in vivo, while preserving their phenotype and functionality. Finally, we have developed the protocol for the GMP-compliant production of NR3C1 KO CMV, BKV, and adenovirus multispecific T cells for clinical use.
Silencing the GR to render CMV-specific T cells resistant to the effect of glucocorticoids has been performed by others using transcription activator-like effector nuclease (TALEN) technology.16 In this work, we used CRISPR-Cas9 technology to target the NR3C1 gene in VSTs. CRISPR-Cas9 has a number of advantages over TALEN, including ease of design for genomic targets, easy prediction regarding off-target sites, and the possibility of modifying several genomic sites simultaneously (multiplexing).17-19 In spite of these advantages, CRISPR-Cas9 carries a number of uncertainties that have complicated its therapeutic translation, including the risk of off-target genome editing20 and challenges in translating this technology safely to the clinic.21,22 In this study, we have addressed these concerns by developing a GMP-compliant protocol for the large-scale expansion and gene editing of VSTs. In addition, we have taken a number of steps to assure the safety of our approach by minimizing the possibility of off-target genetic events. These steps include designing the optimal gRNA based on computational and mathematical predictions and the application of functional experimental off-target cleavage validation assays using GUIDE-seq8 and rhAmp-Seq10 technologies. In addition, we used an RNP complex for Cas9-sg RNA delivery. This approach has been shown to limit off-target events as the RNP degrades rapidly in the cells, limiting its activity.23 Finally, we used GMP-grade high-fidelity Cas9 protein (Aldevron)24 to further minimize genome-wide off-target activities to ensure the safety of our product.
Despite all precautions, it is still possible that off-targets events may lead to unforeseen toxicities such as malignant transformation. We believe that adoptive cellular therapy with off-the-shelf HLA-mismatched VSTs is a good platform to introduce CRISPR-Cas9 gene editing to the clinic.25,26 Non–gene-edited VSTs have a proven track record of safety as reported in multiple clinical trials.1 They are differentiated mature T cells with minimal, if any, potential for malignant transformation. Furthermore, they will eventually be rejected because of the HLA mismatch between the donor and the recipient and in the unlikely event of toxicity, NR3C1 KO VSTs can still be eliminated using antibodies against T cells such as antithymocyte globulin or alemtuzumab (that targets CD52).
To translate our approach to the clinic, we have validated the use of GLP-grade viral PepMix and GMP-grade gRNA and high-fidelity Cas9 protein, and scaled up the transfection protocol to facilitate the generation of clinically relevant numbers of VSTs. We have shown that NR3C1 KO VSTs generated using our GMP-compliant approach have a comparable potency to non–gene-edited VSTs, even when cultured in the presence of glucocorticoids. In addition, in an immunodeficient mouse model, the NR3C1 KO VSTs engrafted could be detected at similar frequencies to non–gene-edited VSTs and were resistant to the lymphocytotoxic effect of dexamethasone. We confirmed the potency of NR3C1 KO VSTs in a series of in vitro experiments because a robust mouse model of human viral infection to test the efficacy of adoptively infused VSTs is not readily available.
Our approach for the large-scale deletion of the GR can be extended to engineer other cellular therapy products to target cancers in which concomitant use of glucocorticoids is often required, such as with brain cancers.27,28 In addition, it can be applied to engineer regulatory T cells to allow their coadministration with steroids for the treatment of GVHD29 or autoimmune disorders.30,31 Indeed, our methodology to silence the expression of genes using CRISPR-Cas9 and to produce large-scale GMP-compliant cells for cell therapy could be used to target a myriad of genes, opening the way for multiple clinical applications.
In summary, we have developed a novel approach for the generation of large numbers of GMP-compliant, steroid-resistant VSTs, targeting CMV, BKV, and adenovirus, that can be used to treat severe viral infections in immunosuppressed patients, irrespective of whether the patient is receiving glucocorticoid therapy. A clinical study to assess the safety and efficacy of this strategy will be conducted in our institution.
Data-sharing requests may be e-mailed to the corresponding author, Katayoun Rezvani, at krezvani@mdanderson.org.
Acknowledgments
This work was supported by the National Institutes of Health, National Cancer Institute (CA061508-21A1 and CA211044-01) and the MD Anderson Cancer Center Acute Myeloid Leukemia (AML) Moon Shot Program.
Authorship
Contribution: R.B., A.A., N.U., E.G., and M.D. performed experiments and interpreted and analyzed the data; M.M., M.H.S., L.N.K., L.L., L.B., and P.P.B. provided advice on experiments and commented on the manuscript; E.E., G.O., A.K.N.C., J.L., S.L., M.K., V.N., M.B., and A.D.K. assisted with experiments; Y.L., E.L., S. Acharya, S. Ang, S.M., D.M., R.C., and E.J.S. commented on the manuscript; M.C.G., R.T., M.S.S., G.R.R., M.S.M., G.K., and M.A.B. performed off-target effect studies and commented on the manuscript; N.W.F. performed pathological examinations on mouse tissue; K.R. designed and directed the study; and M.D., L.M.-F., and K.R. wrote the manuscript.
Conflict-of-interest disclosure: R.T., M.S.S., G.R.R., M.S.M., G.K., and M.A.B. are employed by Integrated DNA Technologies, Inc (IDT), which manufactures reagents similar to some described in the manuscript. M.A.B., M.S.M., R.T., and G.R.R. own equity in DHR, the parent company of IDT. The remaining authors declare no competing financial interests.
Correspondence: Katayoun Rezvani, Department of Stem Cell Transplantation and Cellular Therapy, The University of Texas MD Anderson Cancer Center, 1400 Holcombe Blvd, Houston, TX 77030-4009; e-mail: krezvani@mdanderson.org.

