

Key Points
Nox2 NADPH oxidase is dispensable for platelet ROS generation as well as platelet activation, adhesion, secretion, and aggregation.
Nox2 NADPH oxidase is not essential in mediating experimental thrombosis in major vessels, such as the carotid artery.

Abstract
Deficiency of the Nox2 (gp91phox) catalytic subunit of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a genetic cause of X-linked chronic granulomatous disease, a condition in which patients are prone to infection resulting from the loss of oxidant production by neutrophils. Some studies have suggested a role for superoxide derived from Nox2 NADPH oxidase in platelet activation and thrombosis, but data are conflicting. Using a rigorous and comprehensive approach, we tested the hypothesis that genetic deficiency of Nox2 attenuates platelet activation and arterial thrombosis. Our study was designed to test the genotype differences within male and female mice. Using chloromethyl-dichlorodihydrofluorescein diacetate, a fluorescent dye, as well as high-performance liquid chromatography analysis with dihydroethidium as a probe to detect intracellular reactive oxygen species (ROS), we observed no genotype differences in ROS levels in platelets. Similarly, there were no genotype-dependent differences in levels of mitochondrial ROS. In addition, we did not observe any genotype-associated differences in platelet activation, adhesion, secretion, or aggregation in male or female mice. Platelets from chronic granulomatous disease patients exhibited similar adhesion and aggregation responses as platelets from healthy subjects. Susceptibility to carotid artery thrombosis in a photochemical injury model was similar in wild-type and Nox2-deficient male or female mice. Our findings indicate that Nox2 NADPH oxidase is not an essential source of platelet ROS or a mediator of platelet activation or arterial thrombosis in large vessels, such as the carotid artery.
Introduction
Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a major reactive oxygen species (ROS)–generating enzyme in the vasculature.1 Nox2 (gp91phox) is the transmembrane catalytic subunit of NADPH oxidase that is responsible for the generation of large amounts of ROS from phagocytic cells, such as neutrophils.2 Nox2 is also expressed in platelets3-10 ; however, the role of Nox2-derived ROS in platelet activation, aggregation, and thrombosis remains controversial.5,8,9,11
Early in vitro studies with platelets from healthy humans were the first to suggest that ROS production in platelets and subsequent platelet activation are mediated by NADPH oxidase. Specifically, pharmacological inhibition of NADPH oxidase with the nonspecific inhibitor apocynin or diphenyleneiodonium (DPI) decreased platelet ROS production and platelet activation.6,12-14 The Nox2 catalytic subunit of NADPH oxidase was later suggested to be a mediator of platelet ROS production and activation, based on findings in platelets from patients with chronic granulomatous disease (CGD; an X-linked disease caused by mutations in the gene CYBB, which encodes Nox215 ). Platelets from CGD patients demonstrated diminished agonist-induced generation of ROS.12,16 However, despite a significant decrement in ROS generation, platelet aggregation was not altered in CGD patients,12,16 suggesting that the absence of Nox2 does not influence major platelet functions.
Studies of platelet function in murine models with genetic deficiency of Nox2 are limited and provide conflicting results. Walsh et al8 demonstrated no change in ROS production, platelet activation, ATP release, platelet spreading, or aggregation in platelets from Nox2-deficient mice compared with wild-type (WT) littermates. Likewise, Dharmarajah et al5 observed similar aggregation responses in platelets from WT, Nox2-deficient, and p47phox (a cytoplasmic subunit of NADPH oxidase)–deficient mice. In contrast, Delaney et al9 reported that Nox2-deficient mice exhibit decreased platelet ROS generation, ATP release, aggregation, and in vivo thrombus formation. Therefore, the role of Nox2 in the generation of ROS by platelets and its regulatory role in platelet-dependent thrombosis remain uncertain.
We consider that some of the controversy about the role of Nox2 in platelet ROS generation may be linked to the fact that methods to measure ROS have several limitations and often lack specificity, sensitivity, and reproducibility.17-19 Secondly, the discrepant results in murine models may be related to potential sex differences owing to the X-linked status of the CYBB gene. Therefore, in this study, we adopted a rigorous and comprehensive approach to test the mechanistic role of endogenous Nox2 in platelet function and thrombosis. Our approach included several methods to measure intraplatelet ROS, use of murine and human models of Nox2 deficiency, and comparison of Nox2-deficient male (Cybb−/y) and female (Cybb−/−) mice with their WT male (Cybb+/y) or female (Cybb+/+) counterparts. Our data demonstrate that genetic deficiency of Nox2 does not inhibit platelet ROS generation, activation, aggregation, or adhesion responses or susceptibility to arterial thrombosis in male or female mice. Our data also provide evidence that sources other than Nox2 NADPH oxidase must contribute to ROS generation in platelets.
Methods
Mice
The University of Iowa Animal Care and Use Committee approved all murine experiments. Male Nox2-deficient (Cybb−/y) mice on the C57BL6/J background were obtained from The Jackson Laboratory and were backcrossed to C57BL6/J female mice (The Jackson Laboratory) to generate hemizygous WT male (Cybb+/y) and heterozygous female Nox2-deficient (Cybb+/−) breeders. These breeder mice were then intercrossed to generate male WT (Cybb+/y) and knockout (Cybb−/y) littermates. This breeding strategy also generated female Cybb+/+ and Cybb+/− mice. However, due to the X-chromosome location of the Cybb gene, littermates of female Cybb+/+ and Cybb−/− mice could not be generated from the same breeder pairs. Therefore, male Cybb−/y mice were bred with female Cybb+/− mice, which generated female Cybb−/− and Cybb+/− mice. Genotyping for the WT and targeted Cybb alleles was performed by polymerase chain reaction.9 Mice were studied between 8 and 12 weeks of age.
Human subjects
The study on human subjects was approved by the Institutional Review Board, University of Iowa. All participants gave written informed consent. The diagnosis for X-linked CGD was made by demonstrating the absence of NADPH oxidase activity in neutrophils using a nitroblue tetrazolium test. The diagnosis was confirmed by mutation analysis for the CYBB gene that demonstrated a splice mutation G264A in exon 3 in the first patient. The second patient has a non-sense mutation C469T. The western blots for these patients showed lack of gp91phox and p22phox proteins. These represent loss of function/null mutations.
Platelet preparation
Mice were anesthetized with isoflurane and bled via the retro-orbital plexus20,21 using a heparin-coated capillary tube, and blood was collected into acid-citrate dextrose (ACD; 9:1). Platelets were prepared as described previously with minor modifications.7 Briefly, blood was diluted with modified Tyrode’s buffer (134 mmol/L NaCl, 2.9 mmol/L KCl, 0.34 mmol/L Na2HPO4, 12 mmol/L NaHCO3, 20 mmol/L HEPES, 1.0 mmol/L MgCl2, 5.0 mmol/L glucose, and 0.35% [weight-to-volume ratio] bovine serum albumin, pH 7.35) and centrifuged at 100g for 15 minutes at room temperature. Platelet-rich plasma (PRP) was isolated, prostaglandin E1 (PGE1; 1 µM) was added to PRP, and samples were centrifuged at 800g for 7 minutes to obtain platelet pellets. Pellets were then washed with modified Tyrode’s buffer in the presence of PGE1 (1 µM) and resuspended in modified Tyrode’s buffer. For all quantitative assays, such as high-performance liquid chromatography (HPLC), messenger RNA (mRNA), or protein expression, blood was drawn via carotid artery cannulation into ACD (9:1), washed platelets were prepared as above, and platelets were further purified with CD45 and Ter-119–labeled MicroBeads (Miltenyi Biotec, Auburn, CA), as described.22,23 Briefly, platelet suspensions (2.5 × 108/mL) were incubated with 35 µL of Ter-119 and CD45 for 20 minutes in the dark. Red blood cells and leukocyte-bound beads were separated on a precalibrated MACS column, as per the manufacturer’s instructions. The purity of platelets prepared through this approach was tested in preliminary experiments using immunofluorescence staining for platelets (with DyLight 488–labeled GpIbβ antibody; Emfret) and nucleated cells (with 4′,6-diamidino-2-phenylindole), as reported previously.22,23,24 Manual cell counting in 25 image fields showed no nucleated cells in 105 platelets counted (supplemental Figure 1).
Washed human platelets were prepared as described previously25 with minor modifications. Whole blood from human subjects was collected in 3.2% sodium citrate (9:1, volume-to-volume ratio) and centrifuged at 100g for 15 minutes to obtain PRP. A fraction of PRP was supplemented with EDTA (1 mM) and PGE1 (1 µM) and then centrifuged at 800g for 10 minutes. The supernatant fraction containing platelet-poor plasma was transferred to a separate tube, and pellets were washed in the presence of PGE1 (1 µM) using wash buffer (134 mmol/L NaCl, 2.9 mmol/L KCl, 0.34 mmol/L Na2HPO4, 12 mmol/L NaHCO3, 20 mmol/L HEPES, 1.0 mmol/L MgCl2, 5.0 mmol/L glucose, 1 mM EDTA, pH 6.5) and resuspended into modified Tyrode’s buffer.
Measurement of ROS in platelets using fluorescent dyes
Levels of platelet-derived ROS were measured using fluorescent dye methods.14 Briefly, platelets (2 × 108/mL) were incubated with 10 µM 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (Molecular Probes), a chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate, for 15 minutes at 37°C in the dark. 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate–loaded platelets were then incubated in the presence or absence of thrombin or convulxin for 15 minutes at room temperature. The sample was diluted 10 times with 1× Dulbecco’s phosphate-buffered saline, and the fluorescent signal of the oxidized adduct of chloromethyl derivative of 2′,7′-dichlorofluorescin diacetate (CM-H2DCF; ie, DCF) was measured by flow cytometry. To measure the time course of increases in DCF fluorescence, an alternative experiment was performed: washed platelets loaded with CM-H2DCF were activated with thrombin (0.02 or 0.1 U/mL), and the samples were diluted for different time intervals (2-15 minutes) and analyzed immediately by flow cytometry. Data on geometric mean fluorescence intensity were collected and presented as fold change over unstimulated WT platelets.
Similarly, to probe for mitochondrial ROS, 2 probes (dihydrorhodamine 1,2,3 [DHR1,2,3] and MitoSox) were used at a concentration of 10 µM and incubated with platelets in the presence or absence of thrombin or convulxin for 25 minutes, and the fluorescent signal was measured by flow cytometry. The data on geometric mean fluorescence intensity were collected and presented as fold change over unstimulated WT platelets.
Quantification of platelet-derived superoxide using HPLC
Bead-purified washed murine platelets (pooled, 4 × 108/mL) were incubated with 25 µM dihydroethidium (DHE) for 25 minutes at 37°C, activated or aggregated with thrombin and convulxin for 5 minutes, and centrifuged. To prepare activated platelets, samples were incubated with convulxin (50 ng/mL) + thrombin (0.05 U/mL) without stirring; to prepare aggregated platelets samples were incubated with convulxin (50 ng/mL) + thrombin (0.05 U/mL) while stirring in an aggregometer. Platelet pellets were solubilized26 and analyzed by HPLC coupled with electrochemical detection (ESA CoulArray). The superoxide-specific oxidation product 2-hydroxyethidium (2-OH-E+) and other oxidation products of DHE (also known as hydroethidine [HE]), such as E+ (ethidium), E+ dimer (ethidium dimer), DHE dimer, and HE-E+ (hydroethidine-ethidium dimer), were analyzed.26,27 Potentials on the electrochemical detector(s) were set at 0, 200, 280, 365, 400, 450, and 600 mV. Quantification of oxidation products in platelet pellets, supernatants, and standards was accomplished by measuring the total peak areas in nanocoulombs (nA·s) and the use of appropriate standard curves for each species.
Flow cytometric analysis of integrin αIIbβ3 activation and α-granule secretion
Activation of integrin αIIbβ3 and α-granule secretion were evaluated by quantifying JON/A binding and surface expression of P-selectin, respectively. Washed platelets (2 × 108/mL) were activated with human thrombin or convulxin, incubated with 10 μL of phycoerythrin-conjugated JON/A antibody (1:500) or fluorescein isothiocyanate–conjugated rat anti-mouse CD62P antibody (1:500; both from BD Biosciences) for 15 minutes at 37°C, fixed in 1% paraformaldehyde, and analyzed using a Becton Dickinson LSR II. The data on geometric mean fluorescence intensity were collected and presented as fold-change over unstimulated WT platelets.
Platelet aggregation
Human PRP or washed human/mouse platelets (2 × 108/mL) were stirred (1200 rpm at 37°C) with collagen (0.1-2 μg/mL, CHRONO-LOG), convulxin (25 ng/mL, Santa Cruz Biotechnology), or thrombin (0.02-0.1 U/mL; CHRONO-LOG) in a CHRONO-LOG aggregometer (Model 560-VS). Washed platelets were incubated with CaCl2 (1 mM) for 1 minute prior to aggregation. Aggregation was measured as the percentage change in light transmission, where 100% refers to transmittance through blank solutions (modified Tyrode’s buffer for washed platelets and platelet-poor plasma for PRP).
Microfluidics-based platelet adhesion on collagen matrix under arterial shear
Platelet adhesion and thrombus growth on collagen matrix were measured in a microfluidic BioFlux flow chamber (Fluxion Biosciences). High-shear plates were coated with 50 μg of collagen (CHRONO-LOG) and blocked with 1% bovine serum albumin. Platelets (2 × 108/mL) labeled with calcein green (2.5 µg/mL) were perfused over collagen at a physiological arterial shear rate (2000/s) for 5 minutes. Platelet adherence over time was recorded and analyzed using ImageJ (National Institutes of Health). Thrombus growth over time was measured by accumulation of adhering platelets in a fixed field. Total thrombi area at 5 minutes was calculated by the average accumulation of platelets in 5 representative fields.
Carotid artery thrombosis
Carotid artery thrombosis was induced by photochemical injury, as described previously.28 Briefly, the right common carotid artery was transilluminated with a 1.5-mV, 540-nm green laser (Melles Griot). Rose Bengal (35 mg/kg) was injected via a femoral vein catheter. Carotid artery blood flow was monitored with a Doppler flow probe for up to 90 minutes after injury or until a stable occlusion developed. The stable occlusion was defined as the time at which blood flow remained absent for ≥10 minutes.
Statistical analysis
GraphPad Prism software, version 7.04 was used for statistical analysis. Data from fluorescence-activated cell sorting–based experiments were analyzed using 2-way analysis of variance (ANOVA) with the Tukey test for multiple comparisons. Platelet aggregation data were analyzed using multiple Student t tests with the Holm-Sidak test for comparison. Platelet accumulation over time was compared using 2-way ANOVA with repeated measures, followed by the Tukey test for multiple comparisons. The times to stable occlusion postphotochemical injury were compared using 2-way ANOVA with the Tukey test for multiple comparisons. Other statistical analysis details are provided in the figure legends. Statistical significance was defined as P < .05. Values are reported as mean ± standard error.
Results
mRNA and protein levels of subunits of NADPH oxidase in WT and Nox2-deficient mice
We first confirmed that mRNA for Nox2 is expressed in platelets and lungs from WT mice but is absent in Nox2-deficient mice (supplemental Figure 2A,C). Nox2 deficiency had no effect on platelet mRNA levels of the cytoplasmic subunit, p47phox (supplemental Figure 2B). We were unable to detect expression of Nox1 or Nox4 mRNA in platelets from WT mice, which is in accordance with previous findings.7 Similarly, no expression of Nox1 or Nox4 mRNA was detected in platelets from Nox2-deficient mice (data not shown). Immunoblotting showed similar protein levels of Nox1, Nox4, and p47phox in platelets from WT or Nox2-deficient mice (supplemental Figure 3). Thus, our data indicate that the absence of Nox2 does not alter the expression of other NADPH subunits in platelets.
Deficiency of Nox2 does not alter platelet ROS generation in male or female mice
We used several approaches to measure platelet-derived ROS levels in Nox2-deficient male and female mice and compared them with levels in sex-matched WT mice. First, using the redox-sensitive CM-H2DCFDA dye as a fluorescent probe for ROS, we did not detect any differences in oxidation of this probe between Cybb+/y and Cybb−/y males or between Cybb+/+ and Cybb−/− females at baseline or after activation with 2 agonists (convulxin or thrombin) (Figure 1) for 15 minutes. Furthermore, the time course of ROS generation in the presence of thrombin was similar in both genotypes (supplemental Figure 4).
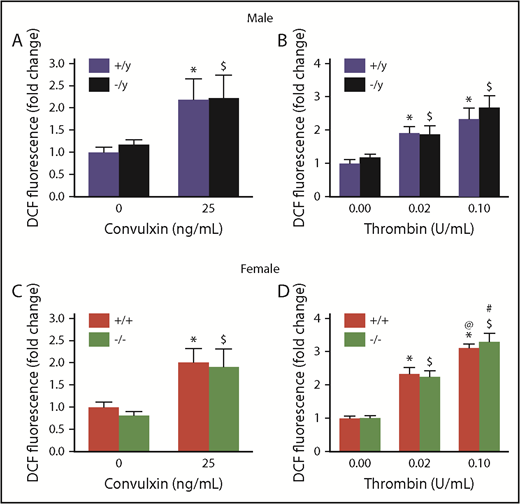
Nox2 deficiency does not alter platelet ROS production in male or female mice. Levels of intracellular ROS, detected by oxidation of CM-H2DCF, were measured in the presence or absence of stimulation with convulxin or thrombin in washed platelets from male (A-B) and female (C-D) WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice. The fluorescent signal generated as the result of oxidation of CM-H2DCF to CM-DCF is presented as fold change over the signal observed with platelets from sex-matched WT mice without thrombin or convulxin. Data are presented as mean ± standard error (n = 6 or 7 mice in each group). *P < .05 vs. WT (+/y or +/+) without convulxin/thrombin, $P < .05 vs. Nox2-deficient (−/y or −/−) without convulxin/thrombin, @P < .05 vs. WT (+/y or +/+) with thrombin (0.02 U/mL), #P < .05 vs. Nox2-deficient (−/y or −/−) with thrombin (0.02 U/mL), 2-way ANOVA with the Tukey test for multiple comparisons.
Nox2 deficiency does not alter platelet ROS production in male or female mice. Levels of intracellular ROS, detected by oxidation of CM-H2DCF, were measured in the presence or absence of stimulation with convulxin or thrombin in washed platelets from male (A-B) and female (C-D) WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice. The fluorescent signal generated as the result of oxidation of CM-H2DCF to CM-DCF is presented as fold change over the signal observed with platelets from sex-matched WT mice without thrombin or convulxin. Data are presented as mean ± standard error (n = 6 or 7 mice in each group). *P < .05 vs. WT (+/y or +/+) without convulxin/thrombin, $P < .05 vs. Nox2-deficient (−/y or −/−) without convulxin/thrombin, @P < .05 vs. WT (+/y or +/+) with thrombin (0.02 U/mL), #P < .05 vs. Nox2-deficient (−/y or −/−) with thrombin (0.02 U/mL), 2-way ANOVA with the Tukey test for multiple comparisons.
We next examined generation of superoxide and hydroxyl radicals by platelets using electron spin resonance (ESR) spin trapping with 5,5-dimethyl-1-pyrroline N-oxide (DMPO). As a control, we first established that neutrophils (2 × 106/mL) from WT mice, but not Nox2-deficient mice, had the anticipated phorbol 12-myristate 13-acetate–induced dose-dependent increase in extracellular superoxide and hydroxyl radical spin adducts and that the ESR signals were completely quenched by superoxide dismutase (supplemental Figure 5A-C). In contrast, platelets from WT and Nox2-deficient mice, at much higher density than neutrophils (4 × 108/mL), did not generate any detectable signals for DMPO-superoxide or hydroxyl radical spin adducts, even when stimulated with convulxin and thrombin together (supplemental Figure 5A,D-E)
Next, we used a highly specific and sensitive HPLC-based method to measure platelet-derived superoxide. The advantage of this approach is that it can distinguish between the oxidation products derived from the oxidation of DHE by superoxide and other oxidation products generated from DHE, including products of DHE auto-oxidation. The 2-OH-E+ signal is the only superoxide-specific product of DHE oxidation; other oxidation products are nonspecific that arise from quite different intracellular chemistry. A representative panel of HPLC tracings for various oxidation products in platelets from WT and Nox2-deficient mice is presented in supplemental Figure 6. We first observed that resting platelets from WT and Nox2-deficient mice exhibit similar amounts of intracellular 2-OH-E+. Upon activation with convulxin and thrombin, platelets from WT and Nox2-deficient mice display no increases in the amount of 2-OH-E+. Under experimental conditions in which convulxin and thrombin produced aggregation, an overall significant elevation in 2-OH-E+ signal was observed in platelets, regardless of the genotype (P < .05 compared with resting platelets), but there were no differences in the amounts of 2-OH-E+ produced by aggregated platelets from WT and Nox2-deficient mice (Figure 2A; Table 1). In contrast, very minimal extracellular 2-OH-E+ was detected with either genotype under these conditions (Figure 2B; Table 1). Other nonspecific DHE oxidation products are shown in Table 1. Control experiments with DHE in buffer only (without platelets) indicate that there is no background oxidation of DHE in the time frame of these experiments (supplemental Table 1). These data indicate that there is no detectable formation of superoxide by platelets from a Nox2 enzyme system.
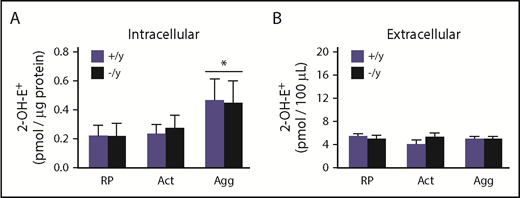
The amounts of superoxide-specific 2-OH-E+generated from the oxidation of DHE are similar in Nox2-deficient and WT mice. Bead-purified platelets (pooled) from male Nox2-deficient (−/y) or WT (+/y) mice were incubated with DHE (25 µM), followed by activation (no stirring) or aggregation (with stirring) with thrombin (0.05 U/mL) and convulxin (50 ng/mL). The superoxide-specific oxidation product 2-OH-E+ was quantitated in platelet pellets (intracellular signal) (A) and supernatant fractions (extracellular signal) (B) using HPLC coupled with electrochemical detection and authentic standards. Data are presented as mean ± standard error (n = 3 or 4 mice in each group). *P < .05 vs. RP. 2-OH-E+ generation within different experimental conditions (ie, RP, Act, or Agg) and genotypes was analyzed using 2-way ANOVA with the Tukey test. Act, activated platelets; Agg, aggregated platelets; RP, resting platelets.
The amounts of superoxide-specific 2-OH-E+generated from the oxidation of DHE are similar in Nox2-deficient and WT mice. Bead-purified platelets (pooled) from male Nox2-deficient (−/y) or WT (+/y) mice were incubated with DHE (25 µM), followed by activation (no stirring) or aggregation (with stirring) with thrombin (0.05 U/mL) and convulxin (50 ng/mL). The superoxide-specific oxidation product 2-OH-E+ was quantitated in platelet pellets (intracellular signal) (A) and supernatant fractions (extracellular signal) (B) using HPLC coupled with electrochemical detection and authentic standards. Data are presented as mean ± standard error (n = 3 or 4 mice in each group). *P < .05 vs. RP. 2-OH-E+ generation within different experimental conditions (ie, RP, Act, or Agg) and genotypes was analyzed using 2-way ANOVA with the Tukey test. Act, activated platelets; Agg, aggregated platelets; RP, resting platelets.
DHE oxidation products in resting, activated, and aggregated platelets
| Nox2 genotype | Thrombin and convulxin | Analyte (pmol)/protein (µg) | ||||
|---|---|---|---|---|---|---|
| 2-OH-E+ | DHE | E+ | E+ dimer | DHE dimer | ||
| Pellet (intracellular) | ||||||
| +/y | — (RP) | 0.24 ± 0.04 | 0.55 ± 0.06 | 1.09 ± 0.05 | 0.24 ± 0.03 | 2.67 ± 0.11 |
| −/y | — (RP) | 0.23 ± 0.06 | 0 | 1.11 ± 0.09 | 0.30 ± 0.06 | 2.03 ± 0.19 |
| +/y | + (Act) | 0.25 ± 0.04 | 0.48 ± 0.06 | 0.80 ± 0.03 | 0.19 ± 0.02 | 1.80 ± 0.10 |
| −/y | + (Act) | 0.28 ± 0.06 | 0.59 ± 0.11 | 0.97 ± 0.07 | 0.12 ± 0.03 | 1.80 ± 0.18 |
| +/y | + (Agg) | 0.47 ± 0.05* | 1.16 ± 0.10 | 1.63 ± 0.07 | 0.68 ± 0.05 | 3.93 ± 0.15 |
| −/y | + (Agg) | 0.45 ± 0.08* | 0.57 ± 0.11 | 1.21 ± 0.09 | 0.47 ± 0.06 | 2.61 ± 0.21 |
| Supernatant (extracellular) | ||||||
| +/y | — (RP) | 5.4 ± 0.8 | 58.6 ± 4.3 | 45.8 ± 2.9 | 2.0 ± 0.7 | 34.0 ± 0.7 |
| −/y | — (RP) | 5.1 ± 1.7 | 57.5 ± 6.9 | 52.0 ± 5.0 | 2.1 ± 1.15 | 33.7 ± 0.3 |
| +/y | + (Act) | 5.2 ± 1.1 | 50.6 ± 5.8 | 38.3 ± 3.8 | 1.8 ± 0.4 | 33.8 ± 0.6 |
| −/y | + (Act) | 5.4 ± 1.6 | 50.9 ± 4.6 | 47.4 ± 5.4 | 1.9 ± 1.0 | 34.0 ± 0.4 |
| +/y | + (Agg) | 5.1 ± 0.7 | 72.0 ± 4.5 | 44.4 ± 2.9 | 1.2 ± 0.7 | 23.0 ± 6.0 |
| −/y | + (Agg) | 5.1 ± 1.1 | 65.0 ± 5.4 | 50.2 ± 4.2 | 3.6 ± 2.3 | 0 |
| Nox2 genotype | Thrombin and convulxin | Analyte (pmol)/protein (µg) | ||||
|---|---|---|---|---|---|---|
| 2-OH-E+ | DHE | E+ | E+ dimer | DHE dimer | ||
| Pellet (intracellular) | ||||||
| +/y | — (RP) | 0.24 ± 0.04 | 0.55 ± 0.06 | 1.09 ± 0.05 | 0.24 ± 0.03 | 2.67 ± 0.11 |
| −/y | — (RP) | 0.23 ± 0.06 | 0 | 1.11 ± 0.09 | 0.30 ± 0.06 | 2.03 ± 0.19 |
| +/y | + (Act) | 0.25 ± 0.04 | 0.48 ± 0.06 | 0.80 ± 0.03 | 0.19 ± 0.02 | 1.80 ± 0.10 |
| −/y | + (Act) | 0.28 ± 0.06 | 0.59 ± 0.11 | 0.97 ± 0.07 | 0.12 ± 0.03 | 1.80 ± 0.18 |
| +/y | + (Agg) | 0.47 ± 0.05* | 1.16 ± 0.10 | 1.63 ± 0.07 | 0.68 ± 0.05 | 3.93 ± 0.15 |
| −/y | + (Agg) | 0.45 ± 0.08* | 0.57 ± 0.11 | 1.21 ± 0.09 | 0.47 ± 0.06 | 2.61 ± 0.21 |
| Supernatant (extracellular) | ||||||
| +/y | — (RP) | 5.4 ± 0.8 | 58.6 ± 4.3 | 45.8 ± 2.9 | 2.0 ± 0.7 | 34.0 ± 0.7 |
| −/y | — (RP) | 5.1 ± 1.7 | 57.5 ± 6.9 | 52.0 ± 5.0 | 2.1 ± 1.15 | 33.7 ± 0.3 |
| +/y | + (Act) | 5.2 ± 1.1 | 50.6 ± 5.8 | 38.3 ± 3.8 | 1.8 ± 0.4 | 33.8 ± 0.6 |
| −/y | + (Act) | 5.4 ± 1.6 | 50.9 ± 4.6 | 47.4 ± 5.4 | 1.9 ± 1.0 | 34.0 ± 0.4 |
| +/y | + (Agg) | 5.1 ± 0.7 | 72.0 ± 4.5 | 44.4 ± 2.9 | 1.2 ± 0.7 | 23.0 ± 6.0 |
| −/y | + (Agg) | 5.1 ± 1.1 | 65.0 ± 5.4 | 50.2 ± 4.2 | 3.6 ± 2.3 | 0 |
Bead-purified platelets (pooled) were incubated with DHE (25 µM), followed by activation (Act; no stirring) or aggregation (Agg; with stirring) with thrombin (0.05 U/mL) and convulxin (50 ng/mL). The oxidation products of DHE, 2-OH-E+, DHE (unreacted probe), E+, E+ dimer, and DHE dimer were quantitated in platelet pellets (intracellular) and supernatant fractions (extracellular). Data are mean ± standard error of the mean (n = 3 or 4 in each group, with 3 technical replicates of each sample).
P < .05 vs. RP for both genotypes.
We next investigated the possibility that the similarity in superoxide signals detected by HPLC between Nox2-deficient and WT platelets might be due to a compensatory increase in platelet mitochondrial ROS in Nox2-deficient platelets. We used 2 probes that accumulate within mitochondria: MitoSOX, a mitochondrially targeted probe with chemistry parallel to DHE, and DHR1,2,3, which can be oxidized by superoxide, hydroxyl radicals, and reactive nitrogen species. DHR1,2,3 (Figure 3A-B) and MitoSOX (Figure 3C-D) generated similar fluorescent signals in platelets from WT or Nox2-deficient male and female mice at baseline or upon activation with different doses of convulxin or thrombin. These data indicate that the absence of Nox2 does not result in compensatory changes in mitochondrial ROS production.
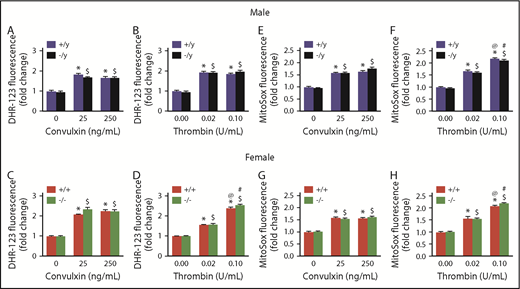
Mitochondrial ROS generation is not altered in Nox2-deficient mice. Washed platelets from male and female mice deficient in Nox2 (−/y or −/−) or WT littermates (+/y or +/+) were incubated with 10 µM of DHR1,2,3 (A-D) or MitoSox (E-H) for 25 minutes at 37°C, in the presence or absence of stimulation with convulxin or thrombin. Samples were analyzed by flow cytometry. Data are presented as mean ± standard error (n = 4 or 5 mice in each group). *P < .05 vs. WT (+/y or +/+) without convulxin/thrombin, $P < .05 vs. Nox2-deficient (−/y or −/−) without convulxin/thrombin, @P < .05 vs. WT (+/y or +/+) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), #P < .05 vs. Nox2-deficient (−/y or −/−) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), 2-way ANOVA with the Tukey test for multiple comparisons.
Mitochondrial ROS generation is not altered in Nox2-deficient mice. Washed platelets from male and female mice deficient in Nox2 (−/y or −/−) or WT littermates (+/y or +/+) were incubated with 10 µM of DHR1,2,3 (A-D) or MitoSox (E-H) for 25 minutes at 37°C, in the presence or absence of stimulation with convulxin or thrombin. Samples were analyzed by flow cytometry. Data are presented as mean ± standard error (n = 4 or 5 mice in each group). *P < .05 vs. WT (+/y or +/+) without convulxin/thrombin, $P < .05 vs. Nox2-deficient (−/y or −/−) without convulxin/thrombin, @P < .05 vs. WT (+/y or +/+) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), #P < .05 vs. Nox2-deficient (−/y or −/−) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), 2-way ANOVA with the Tukey test for multiple comparisons.
Together, these findings suggest that, under physiological conditions, generation of ROS within platelets does not require Nox2.
Deficiency of Nox2 does not alter platelet activation, aggregation, or adhesion in male or female mice
To investigate the role of Nox2 in platelet function in vitro, we first examined agonist-induced activation of integrin αIIbβ3 and P-selectin expression in platelets. Nox2 deficiency in either sex did not alter activation of αIIbβ3 or expression of P-selectin in response to low or high doses of thrombin or convulxin (Figure 4). Similarly, platelet aggregation in response to thrombin or convulxin was not altered with Nox2 deficiency in male or female mice (Figure 5A-F). Furthermore, release of ATP from platelet-dense granules was similar in both genotypes (supplemental Figure 7).
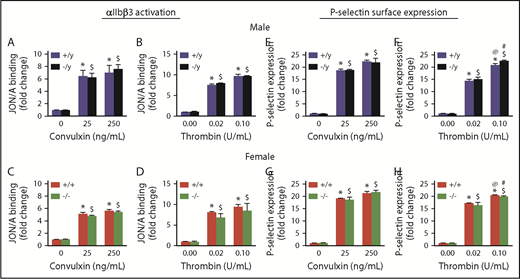
Activation of αIIbβ3and P-selectin surface expression are not influenced by Nox2 deficiency in male or female mice. Activation of αIIbβ3 (detected by JON/A binding) (A-D) and surface expression of P-selectin (E-H) were measured by flow cytometry in washed platelets from male and female WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice following activation with convulxin or thrombin. Data are presented as mean ± standard error (n = 5 or 6 mice in each group). *P < .05 vs. WT (+/y or +/+) without convulxin/thrombin, $P < .05 vs. Nox2-deficient (−/y or −/−) without convulxin/thrombin, @P < .05 vs. WT (+/y or +/+) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), #P < .05 vs. Nox2-deficient (−/y or −/−) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), 2-way ANOVA with Tukey test for multiple comparisons.
Activation of αIIbβ3and P-selectin surface expression are not influenced by Nox2 deficiency in male or female mice. Activation of αIIbβ3 (detected by JON/A binding) (A-D) and surface expression of P-selectin (E-H) were measured by flow cytometry in washed platelets from male and female WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice following activation with convulxin or thrombin. Data are presented as mean ± standard error (n = 5 or 6 mice in each group). *P < .05 vs. WT (+/y or +/+) without convulxin/thrombin, $P < .05 vs. Nox2-deficient (−/y or −/−) without convulxin/thrombin, @P < .05 vs. WT (+/y or +/+) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), #P < .05 vs. Nox2-deficient (−/y or −/−) with convulxin (25 ng/mL)/thrombin (0.02 U/mL), 2-way ANOVA with Tukey test for multiple comparisons.
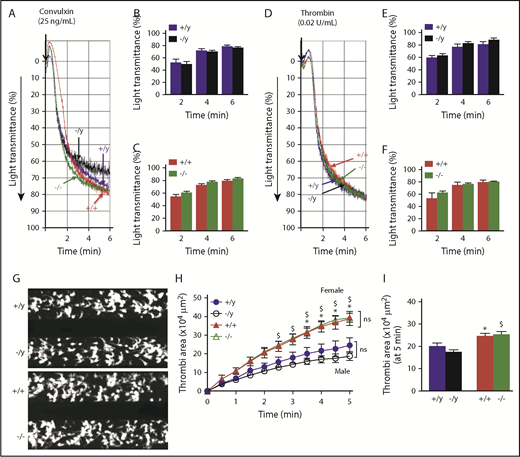
Platelet aggregation and accumulation/thrombi formation ex vivo are similar in WT and Nox2-deficient male or female mice. Washed platelets from male and female WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice were activated with 25 ng/mL convulxin (A-C) or 0.02 U/mL thrombin (D-F). Representative aggregation tracings for convulxin (A) and thrombin (D). Quantitative data for the percentage light transmission at different time points for convulxin in male (B) and female (C) mice and for thrombin in male (E) and female (F) mice. Washed platelets from male and female WT and Nox2-deficient mice were perfused over a collagen surface for 5 minutes in a microfluidic flow chamber at a shear rate of 2000/s. (G) Representative images of platelet accumulation after 5 minutes of perfusion for male and female mice. (H) The time course of accumulation of platelets/thrombi development was calculated as the surface area covered by platelets in a fixed field. (I) Total thrombi area after 5 minutes of perfusion was calculated as the average surface area covered by platelets in 5 representative fields. Data are presented as mean ± standard error (n = 5-7 mice in each group). Platelet aggregation data were analyzed using multiple Student t tests with Holm-Sidak test for comparison between the groups. Platelet accumulation over time was compared using 2-way ANOVA repeated measures, and total thrombi areas on collagen surface were analyzed by 1-way ANOVA, followed by the Tukey test for multiple comparisons. *P < .05 vs. +/y, $P< .01 vs. −/y.
Platelet aggregation and accumulation/thrombi formation ex vivo are similar in WT and Nox2-deficient male or female mice. Washed platelets from male and female WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice were activated with 25 ng/mL convulxin (A-C) or 0.02 U/mL thrombin (D-F). Representative aggregation tracings for convulxin (A) and thrombin (D). Quantitative data for the percentage light transmission at different time points for convulxin in male (B) and female (C) mice and for thrombin in male (E) and female (F) mice. Washed platelets from male and female WT and Nox2-deficient mice were perfused over a collagen surface for 5 minutes in a microfluidic flow chamber at a shear rate of 2000/s. (G) Representative images of platelet accumulation after 5 minutes of perfusion for male and female mice. (H) The time course of accumulation of platelets/thrombi development was calculated as the surface area covered by platelets in a fixed field. (I) Total thrombi area after 5 minutes of perfusion was calculated as the average surface area covered by platelets in 5 representative fields. Data are presented as mean ± standard error (n = 5-7 mice in each group). Platelet aggregation data were analyzed using multiple Student t tests with Holm-Sidak test for comparison between the groups. Platelet accumulation over time was compared using 2-way ANOVA repeated measures, and total thrombi areas on collagen surface were analyzed by 1-way ANOVA, followed by the Tukey test for multiple comparisons. *P < .05 vs. +/y, $P< .01 vs. −/y.
We next examined adhesion of platelets under arterial shear stress on a collagen surface over 5 minutes by measuring the surface area covered by accumulating platelets: representative images are presented in Figure 5G-H. By monitoring a single field over time, we observed that platelets from male or female mice deficient in Nox2 accumulated at rates comparable to platelets from sex-matched WT mice, indicating similar kinetics of thrombi development (Figure 5I). The total area covered by platelets after 5 minutes of perfusion did not demonstrate a genotype effect in male or female mice (Figure 5J). Interestingly, we observed that female mice developed larger thrombi than male mice regardless of the genotype, suggesting that female mice have a greater potential to develop platelet thrombi under arterial shear stress.
Together, these findings suggest that deficiency of Nox2 in male or female mice is not sufficient to decrease platelet activation, aggregation, and adhesion responses.
Platelet function is not impaired in CGD patients
To determine whether platelet function is also unaffected by Nox2 deficiency in humans, we studied platelet samples from 2 male CGD patients (23 and 26 years of age) carrying a mutation in gp91phox. One patient donated a blood sample 2 times, so we present composite data from 3 separate runs for each assay. A control sample was included with every run using platelets from age-matched healthy male volunteers.
In line with our findings in mice, aggregation of washed human platelets in response to collagen or thrombin was not different between control and CGD patients (Figure 6A-D). We also measured aggregation in PRP to consider circulating factors that may be altered in CGD patients and potentially affect platelet responses. The aggregation responses in PRP from CGD and control samples remained identical when stimulated with different doses of collagen (Figure 6E-G). Finally, using the platelet-adhesion assay in a flow chamber, we found that loss of Nox2 in human platelets did not affect total platelet accumulation or the kinetics of platelet thrombi development (Figure 6I-K). Although the sample sizes are small, these findings substantiate our observations in mice that Nox2 is not required for platelet activation or aggregation.
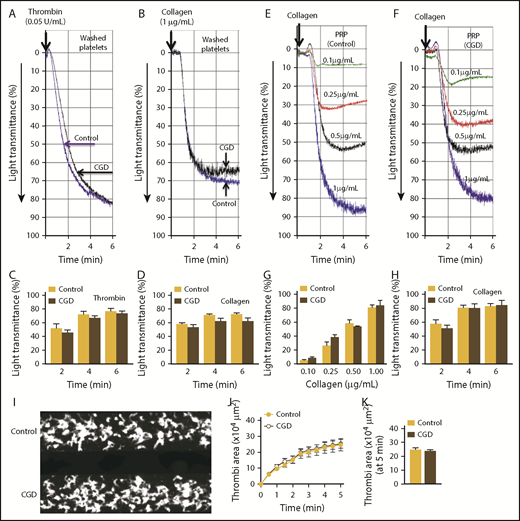
Platelets from CGD patients have similar aggregation and adhesion responses as platelets from healthy control subjects. Platelet aggregation was examined using washed platelets (A-D) or PRP (E-H). Representative aggregation tracings for washed platelets stimulated with thrombin (0.05 U/mL; A) or collagen (1.0 μg/mL; B). Summary data for aggregation kinetics with thrombin (C) or collagen (D). Representative tracings for dose-dependent platelet aggregation to collagen with PRP from control subjects (E) and CGD patients (F). Summary data for maximal aggregation (G) and aggregation kinetics (H) with 1.0 µg/mL collagen of platelets from control subjects vs. CGD patients. Accumulation of platelet thrombi over collagen was examined in a microfluidic flow chamber at a shear rate of 2000/s. (I) Representative images of platelet accumulation/thrombi formation with platelets from control subjects vs. CGD patients. (J) Time course of accumulation of platelets/thrombi development, calculated as the surface area covered by platelets in a fixed field. (K) Total thrombi area after 5 minutes of perfusion, calculated as the average surface area covered by platelets in 5 representative fields. Composite data are presented from 3 separate runs (2 CGD patients and 3 age-matched control subjects; 1 CGD patient donated a blood sample 2 times).
Platelets from CGD patients have similar aggregation and adhesion responses as platelets from healthy control subjects. Platelet aggregation was examined using washed platelets (A-D) or PRP (E-H). Representative aggregation tracings for washed platelets stimulated with thrombin (0.05 U/mL; A) or collagen (1.0 μg/mL; B). Summary data for aggregation kinetics with thrombin (C) or collagen (D). Representative tracings for dose-dependent platelet aggregation to collagen with PRP from control subjects (E) and CGD patients (F). Summary data for maximal aggregation (G) and aggregation kinetics (H) with 1.0 µg/mL collagen of platelets from control subjects vs. CGD patients. Accumulation of platelet thrombi over collagen was examined in a microfluidic flow chamber at a shear rate of 2000/s. (I) Representative images of platelet accumulation/thrombi formation with platelets from control subjects vs. CGD patients. (J) Time course of accumulation of platelets/thrombi development, calculated as the surface area covered by platelets in a fixed field. (K) Total thrombi area after 5 minutes of perfusion, calculated as the average surface area covered by platelets in 5 representative fields. Composite data are presented from 3 separate runs (2 CGD patients and 3 age-matched control subjects; 1 CGD patient donated a blood sample 2 times).
Alternative sources of platelet ROS
There is substantial evidence that ROS are necessary for platelet activation and adhesion,13,29-33 and Nox2 NADPH oxidase is a major mediator,9,34 yet our findings using Nox2-deficient platelets from mice and humans challenge this paradigm. We hypothesized that alternative sources of ROS within platelets are important for platelet activation. We studied aggregation and adhesion responses in healthy human platelets in the presence of pharmacological inhibitors of alternative oxidative pathways. We found that N-acetyl cysteine (NAC; a cysteine donor) significantly inhibited platelet aggregation in a dose-dependent manner (supplemental Figure 8A-B) and diminished platelet accumulation (supplemental Figure 9A-B), suggesting that these platelet functions are dependent on redox status. Next, we measured platelet aggregation and thrombi formation in the presence of MitoTEMPO (mitochondrial superoxide scavenger) or L-NAME (nitric oxide synthase [NOS] inhibitor); both independently decreased platelet aggregation responses to thrombin in a dose-dependent manner (supplemental Figure 8C-F) and significantly inhibited accumulation of platelets over collagen (supplemental Figure 9C-F). Further, an additive effect of these inhibitors was observed on platelet aggregation (supplemental Figure 8G-H) and adhesion (supplemental Figure 9G-H). These data suggest that ROS derived from multiple sources, such as mitochondria and NOS, contribute to platelet aggregation and adhesion. We also observed similar inhibitory effects of NAC, MitoTEMPO, and L-NAME on platelet aggregation (supplemental Figure 10A-C) and thrombi formation (supplemental Figure 10D-F) in platelets from WT or Nox2-knockout mice.
Absence of Nox2 does not decrease susceptibility to arterial thrombosis
To determine the effect of Nox2 deficiency on susceptibility to arterial thrombosis in vivo, we measured the time to occlusion of the carotid artery in response to a photochemical injury in mice. The time to develop a stable occlusion was similar in male and female Nox2-deficient mice compared with sex-matched WT mice (Figure 7). These data suggest that a global loss of Nox2 does not protect mice from arterial thrombosis in major vessels, such as the carotid artery.
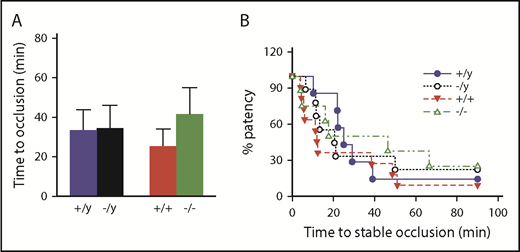
Deficiency in Nox2 does not influence susceptibility to carotid artery thrombosis in male or female mice. The time to stable occlusion of the carotid artery following photochemical injury was measured in male and female WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice. (A) Time to stable occlusion. (B) Percentage of mice with a patent carotid artery (free of stable occlusion) as a function of time after injury. Data are presented as mean ± standard error (n = 7-10 mice in each group). Data were analyzed using 2-way ANOVA with the Tukey test for multiple comparisons.
Deficiency in Nox2 does not influence susceptibility to carotid artery thrombosis in male or female mice. The time to stable occlusion of the carotid artery following photochemical injury was measured in male and female WT (+/y or +/+) or Nox2-deficient (−/y or −/−) mice. (A) Time to stable occlusion. (B) Percentage of mice with a patent carotid artery (free of stable occlusion) as a function of time after injury. Data are presented as mean ± standard error (n = 7-10 mice in each group). Data were analyzed using 2-way ANOVA with the Tukey test for multiple comparisons.
Discussion
Nox2 NADPH oxidase is an important generator of ROS in phagocytes and vascular smooth muscle cells, but its role in platelet ROS production and activation has generated significant controversy over the last 20 years. Several publications have suggested that Nox2 NADPH oxidase is essential for platelet ROS generation and subsequent platelet activation and aggregation.6,9,12-14 Investigators have even proposed developing targeted therapies toward platelet Nox2 NADPH oxidase to prevent platelet hyperactivity.9,35-37 However, some studies in Nox2-deficient platelets from mouse or humans have raised questions about the role of platelet Nox2 NADPH oxidase–derived ROS in platelet activation or aggregation.5,8,11,12,16 To resolve this controversy, we sought to take a comprehensive approach by applying rigorous methods to detect specific ROS and characterize platelet-activation responses in Nox2-deficient murine and human platelets.
Our results herein challenge the paradigm that Nox2 NADPH oxidase is the principal source of ROS responsible for platelet activation and platelet thrombi formation. Unlike prior studies, we used a comprehensive approach to examine the effects of Nox2 on platelet ROS that included (1) a murine model with genetic deficiency in Nox2, as well as human samples from CGD patients, (2) a comparison of platelet ROS generation, activation, aggregation, adhesion, and in vivo thrombosis in Nox2-deficient male and female mice with their respective WT counterparts, (3) utilization of several complementary methods to measure specific ROS in various cellular compartments, and (4) the use of multiple platelet agonists at similar doses across all the assays that yield a better perspective for comparing different responses within platelets. The data generated using these rigorous approaches lead us to conclude that Nox2 is not a mediator of platelet ROS generation, activation, adhesion, or aggregation, and Nox2 does not contribute to thrombosis in large vessels, such as the carotid artery, in male or female mice. Our findings confirm that, although platelet aggregation and adhesion are ROS dependent, the ROS that mediate platelet functional responses are likely generated from other sources, such as mitochondria and NOS, rather than Nox2 NADPH oxidase.
When designing this study, we considered several potential limitations of the previously reported work that could have contributed to the discrepant findings. First, methods to measure ROS have several limitations and often lack specificity, sensitivity, and reproducibility.17-19,38 For example, H2DCFDA is a commonly used probe to detect platelet ROS, but it has limited specificity; several studies have reported the DCF fluorescent signal as a measure of platelet superoxide, which is misleading given that the redox active form of H2DCFDA (H2DCF) does not react directly with superoxide or H2O2 but requires redox active iron to generate more reactive ROS,7,17 Similarly, some previous studies16,34,39 have used luminescent probes, such as lucigenin or the luminol analog L-012, to detect platelet-derived ROS; the validity of these methods has been questioned because the probes themselves can generate superoxide.18 The use of antioxidant enzymes, such as superoxide dismutase or catalase, can improve the specificity of assays that use luminescent probes, but these approaches are often absent in studies of platelet ROS and downstream effects.16,34,39 Second, previous studies with human platelets have often relied on the use of nonspecific pharmacological inhibitors, such as DPI or apocynin, to define the mechanistic contribution of NADPH oxidase to the generation of platelet ROS. However, both of these inhibitors have problems with specificity; DPI inhibits several oxidative enzymes, including NOS, and recent findings suggest that apocynin also lacks specificity for NADPH oxidase.5,40 Therefore, given the limited sensitivity/specificity of the probes and inhibitors, the use of >1 method to measure ROS in the presence of relevant inhibitors is essential.18 Third, because Nox2 is a sex-linked gene, potential differences between males and females may confound studies utilizing Nox2-deficient mice. To overcome these limitations, we used a multidimensional approach for measuring potential ROS generated by platelets, including (1) a chloromethyl derivative of H2DCFDA (CM-H2DCFDA), which has better retention in live cells than carboxy-H2DCFDA used in other studies,9 (2) EPR spin trapping with DMPO, and (3) a highly sensitive and specific HPLC-based method to detect superoxide. Our data demonstrate that Nox2 is not a significant mediator of ROS generation in platelets from male or female mice.
Because some prior studies have reported functional defects in Nox2-deficient platelets,9,12,16 we considered the possibility that Nox2 may support platelet-activation responses independently of ROS generation. Using low and high doses of thrombin and convulxin, we observed that Nox2 deficiency did not alter platelet αIIbβ3 activation or P-selectin expression in either sex. Similarly, there were no alterations in the accumulation of murine or human Nox2-deficient platelets over a collagen surface under arterial shear stress. The discrepant findings between our studies and these prior studies might be explained, in part, by the use of nonspecific pharmacological inhibitors to block NADPH oxidase–mediated platelet activation, which we and other investigators have questioned.5,11 Our findings related to platelet ROS production and platelet activation also differ from a recent report by Delaney et al,9 who used the same Nox2-deficient mouse strain as in our study. We initially questioned whether these differences were potentially due to the fact that Delaney et al appeared to study only female mice, denoted as Nox2−/− mice, in their publication. Also, because control mice were denoted as WT, it is unclear whether the control mice used in their study were Cybb+/+ females generated in the same colony or WT male/female mice from another colony. To avoid potential sex/control bias in our study, we systematically compared findings in male and female mice with their respective sex-matched controls (ie, male Cybb+/y littermates or Cybb+/+ female mice). Using this rigorous approach, we did not detect any Nox2 genotype effects on platelet ROS production, activation, secretion, aggregation, or adhesion in male or female mice. A similar observation was made while comparing aggregation and adhesion responses between platelets from CGD patients and healthy control subjects. We acknowledge that having access to only 2 CGD patients in our study is a limitation; this also has been a limitation of prior studies with CGD patients.16 Overall, our findings are consistent with 2 other studies in Nox2-deficient murine platelets,5,8 as well as previous reports on platelets from CGD patients,12,16 and provide strong support for our conclusion that Nox2 is dispensable for platelet ROS production and function.
We also made 2 additional interesting and novel observations in this study. First, although our study was designed to interrogate the effects of the Nox2 genotype within groups of male or female mice, we also observed an interesting dimorphic effect of sex on susceptibility to thrombi formation. Compared with platelets from male mice, female mice had significantly higher susceptibility to ex vivo platelet thrombi formation, irrespective of Nox2 genotype. Although these observations need to be confirmed in future studies, they are intriguing in light of emerging studies implicating sex differences in platelet activation and associated cardiovascular risks (Framingham Heart Study).41 A second intriguing observation is the significant elevation in superoxide levels in aggregated platelets compared with resting platelets, regardless of Nox2 genotype. These data suggest that superoxide is generated as a downstream response to outside-in-signaling via αIIbβ3. This novel observation warrants examination of oxidative pathways downstream of αIIbβ3 activation in the future that may provide unique targets to control platelet aggregation.
Our findings do not support a role for Nox2 in thrombosis in large arteries, such as the carotid artery. It remains possible that Nox2 may be important for thrombosis in smaller arteries.9 The interplay of hematopoietic and vessel wall factors should be considered in future work, with a focus on platelet-specific deletion of Nox2.
The finding that Nox2 NADPH oxidase does not produce ROS in activated platelets suggests that alternative sources of ROS are important for platelet function. Therefore, we considered the role of alternative oxidative pathways in mediating platelet adhesion and aggregation. We first tested whether loss of Nox2 leads to the compensatory upregulation of other NADPH oxidase subunits within platelets. Our data indicate that genetic deletion of Nox2 does not induce mRNA expression for Nox1 and Nox4 isoforms or upregulate mRNA for p47phox in platelets. Nox2 deletion also did not upregulate protein levels of Nox1, Nox4, or p47phox in platelets. Using platelets from healthy volunteers, we found that the thiol donor and antioxidant NAC inhibits aggregation and adhesion responses, confirming that activation of an oxidative pathway is required for these platelet functions.39,42 The inhibitors of ROS-generating pathways, such as L-NAME and MitoTEMPO, also inhibited platelet aggregation and platelet thrombi formation from human platelets. We found that inhibition of these individual ROS-generating pathways had similar inhibitory effects in WT and Nox2-knockout murine platelets with regard to aggregation (supplemental Figure 10C) and adhesion (supplemental Figure 10F) responses. These findings suggest that Nox2 deficiency is not compensated for by activation of any of these specific oxidative pathways.
Overall, our findings are consistent with prior studies demonstrating a role for ROS in platelet function5,8,43,44 and further suggest that multiple oxidative pathways are involved in regulating platelet aggregation and thrombi formation. These observations are also in concordance with recent reports implicating mitochondrial ROS in platelet activation in certain disease states.45,46 Similarly, a direct role for NOS as a mediator of platelet aggregation has been documented.47,48 A role for p66Shc, a pro-oxidant protein, has also been documented in platelet aggregation.49 It is likely that some of these pathways may remain inactive under physiological conditions but become activated in disease states.45,46,49 Therefore, future studies characterizing specific redox pathways and downstream targets in health and disease are needed to identify the best molecular targets for antioxidant antiplatelet drugs.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Kristina W. Thiel for assistance with manuscript editing.
This work was supported by the National Institutes of Health (NIH), National Institute on Aging (AG049784) (S.D.); a University of Iowa pilot Dance Marathon award (A.A.S. and S.D.); Office of Research and Development, Department of Veterans Affairs grant 2I01BX001729; NIH, National Heart, Lung, and Blood Institute (HL130039) (F.J.M.); and NIH, National Cancer Institute (CA169046 [G.R.B.] and P30CA086862).
Authorship
Contribution: V.K.S. designed the experiments, interpreted the results, and cowrote the manuscript; R.K., M.J., and B.A.W. conducted the experiments; A.A.S., and M.F provided assistance with human subjects; G.R.B. assisted with data analysis; G.R.B, F.J.M., and S.R.L. assisted with the interpretation of the results and cowrote the manuscript; S.D. directed the project, designed the experiments, interpreted the results, and wrote the manuscript; and all authors assisted with the preparation and editing of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sanjana Dayal, Department of Internal Medicine, The University of Iowa Carver College of Medicine, 3160 Med Labs, 200 Hawkins Dr, Iowa City, IA 52242; e-mail: sanjana-dayal@uiowa.edu.

