

Key Points
Nrf2 activation in myeloid cells protects tissues against the deleterious effect of free heme.
Endothelial or myeloid cell–specific responses to Nrf2 activation are distinctive, and both improve the SCD phenotype.

Abstract
Sickle cell disease (SCD) is caused by a monogenic mutation of the β-globin gene and affects millions of people worldwide. SCD is associated with sustained hemolytic anemia, vasoocclusion, ischemia-reperfusion injury, oxidative tissue damage, inflammatory cell activation, and systemic endothelial dysfunction. The transcription factor Nrf2 coordinates the expression of a wide variety of genes encoding antioxidant, detoxification, and metabolic enzymes. Nrf2 participates in suppressing proinflammatory cytokines and organ protection in SCD. However, little is known regarding the mechanisms by which Nrf2 ameliorates SCD pathology or how some cells respond to Nrf2 stimuli to alleviate SCD pathology. Here, we asked whether monocytes/granulocytes and/or endothelial cells are particularly critical in alleviating the pathology of SCD. By targeting these cells with a Cre recombinase system, we generated SCD::Keap1F/F::LysM-Cre and Tie1-Cre mice with constitutive Nrf2 activation in monocytes/granulocytes and endothelial cells, respectively. Analyses of SCD::Keap1F/F::LysM-Cre and SCD::Keap1F/F::Tie1-Cre mice revealed significantly reduced inflammation, along with decreased white blood cell counts and lower Tnfα and Il1β expression in the lungs. Notably, SCD::Keap1F/F::LysM-Cre mice exhibited reduced heme distribution in the liver, consistent with a decrease in the damaged areas. Vascular function in SCD::Keap1F/F::Tie1-Cre mice was significantly improved, with a 50% decrease in vascular leakage and low expression of the adhesion molecules Vcam1 and P-selectin. Thus, Nrf2 activation in monocytes/granulocytes and endothelial cells contributes differentially and cooperatively to the improvement of SCD pathology.
Introduction
Sickle cell disease (SCD) is a blood disorder caused by a monogenic mutation that affects millions of people worldwide. The number of SCD cases is highest in Sub-Saharan Africa, including ∼300 000 newborns each year, with a prevalence of 2% to 10%.1 SCD is caused by a single amino acid substitution (glutamine to valine) in the gene encoding β-globin, which is responsible for the production of abnormal hemoglobin S. The disorder is characterized by hemoglobin polymerization, which distorts the shape of red blood cells (RBCs) into a crescent shape (or sickle shape) when the oxygen supply is insufficient.2,3 SCD is a complex disorder involving unique pathological events. First, cellular components, including highly adherent and hyperreactive cells, such as leukocytes, erythrocytes, monocytes, and endothelial cells (ECs), are increased or activated in response to inflammatory stimuli in thrombosis-hemolysis–damaged regions, due in part to the deleterious and inflammatory effect of free heme. Second, the vascular component is characterized by an imbalanced and progressive vasoactive state mediated by decreased nitric oxide synthesis associated with the release of reactive oxygen species (ROS). Recurrent cycles of ischemia-reperfusion sustain the release of ROS, further facilitating an inflammatory environment.
Activation of the transcription factor nuclear factor erythroid–derived 2-like 2 (Nrf2) can protect tissues or organs in various mouse lines that serve as human disease models. For instance, Nrf2 protects the mouse liver from acetaminophen-induced toxicity, attenuates lung inflammation,4,5 and counteracts ROS-induced organ injury.6,7 Importantly, a recent study identified an ROS-independent mechanism by which Nrf2 expression in bone marrow–derived macrophages represses inflammation.8 The development of a humanized SCD mouse model9,10 has facilitated comprehensive studies of the physiopathology and complications of the disease, leading to the establishment of novel therapeutic approaches to treat SCD.11 We recently demonstrated that the use of 2-cyano-3,12-dioxooleana-1, 9(11)-dien-28-oic acid imidazolide (a triterpenoid chemical that induces the transcription factor Nrf2) and genetic activation of Nrf2 by knocking down the Kelch like-ECH-associated protein 1 (Keap1) gene mitigates SCD complications in mice.12 Nrf2 activation in SCD mice impairs the progression of inflammation by reducing the white blood cell (WBC) count and the expression levels of proinflammatory cytokine genes, including interleukin-6 (IL6) and interleukin-1β (IL1β). Moreover, Nrf2 activation rescues liver necrosis and ameliorates plasma heme clearance.
Nrf2 is a master transcription factor that mediates the gene expression of a variety of detoxifying and antioxidative enzymes/proteins.13,14 Cellular Nrf2 activity is controlled mainly by Keap1, a substrate adaptor protein of the Cullin-3 (Cul3)–based E3 ubiquitin ligase.15 Under unstressed conditions, Nrf2 is efficiently ubiquitinated by the Keap1-Cul3 ubiquitin ligase complex and is rapidly degraded through the proteasome pathway.16,17 In contrast, upon exposure to oxidative or xenobiotic stressors, the ubiquitin ligase activity of Keap1 is inactivated, and newly generated Nrf2 accumulates within cells by escaping cytoplasmic Keap1-mediated degradation, which is known as the Keap1 floodgate.18 Under these conditions, Nrf2 efficiently translocates into the nucleus, where it heterodimerizes with one of the small Maf proteins and activates the transcription of a variety of cytoprotective enzymes.
In the context of SCD, a few lines of evidence suggest that the activation of Nrf2 target genes involved in the heme/hemoglobin and iron-scavenging machinery, such as ferritin (Ftl1) and hemopexin, can mitigate hemolysis-related SCD pathology.19,20 However, in this regard, a number of key questions remain, such as whether it is necessary to modulate Nrf2 activity in all cells or if there is a crucial cell type responsible for the response of Nrf2-mediated reperfusion-damaged areas to Nrf2 activation in vivo to contribute to the improvement of SCD pathology.
Among the many types of cells that may participate in the establishment of SCD pathology, we focused on ECs and myeloid lineage cells in this study, because these cell types reside near ischemia reperfusion–damaged areas and heme metabolism centers, respectively. In fact, macrophages participate in the clearance of plasma heme/hemoglobin, while ECs manage hemolysis stress. We deleted the Keap1 gene in SCD mice to activate Nrf2 specifically in myeloid lineage cells and ECs. This study revealed that Nrf2 activation in myeloid lineage cells attenuates inflammation and protects the liver against avascular necrosis. In addition to promoting heme clearance from the circulation, Nrf2 activation in myeloid lineage cells prevents the tissue accumulation of toxic heme and iron and promotes heme degradation and iron elimination in organs. Nrf2 activation in ECs protects tissues and cells from heme extravasation, reinforces the integrity of the vascular endothelium, and upregulates the expression of genes encoding scavenging proteins and antioxidant enzymes. These results unequivocally demonstrate that to protect tissues from SCD pathology, Nrf2 activation is required in both myeloid lineage cells and ECs in a distinct but overlapping manner.
Materials and methods
Mice
The Animal Care and Use Committee of Tohoku University approved all animal experiments. We used both male and female homozygous SCD model (hα/hα, βS/βS) mice generated by Townes and colleagues9 and Keap1 flox (Keap1F/F) mice.21 Deletion of the floxed Keap1 allele in myeloid cells or ECs was achieved by crossing Keap1F/F mice with mice harboring Cre recombinase under the regulation of the lysozyme M (LysM) locus21 or Tie1-Cre,22 respectively. The mice were maintained on a mixed background, and 8- to 12-week-old mice were used for the experiments.
Peritoneal cell collection
Nonelicited macrophages were collected from 8-week-old mice via intraperitoneal lavage with phosphate-buffered saline and cultured in RPMI 1640 medium containing 10% fetal bovine serum and 1% penicillin-streptomycin. Cells were incubated at 37°C in 5% CO2 for 4 hours. Adherent cells were washed with phosphate-buffered saline and harvested for experiments.
Plasma biochemistry
Plasma levels of alanine aminotransferase (ALT) and total and direct bilirubin were measured with a Fuji DRI-CHEM 7000 biochemical autoanalyzer (Fujifilm). The level of indirect bilirubin was calculated from the levels of total and direct bilirubin. Total plasma heme levels were measured calorimetrically at 400 μm using a Quanti-Chrome Heme Assay Kit (BioAssay Systems).
Histological analysis
Livers and lungs were fixed in Mildform 10N (Wako Pure Chemical Industries) and processed into paraffin-embedded tissue sections. Lung sections were subjected to hematoxylin and eosin staining, F4/80 staining with a rat anti-mouse F4/80 antibody (Cl: A3-1, AbD Serotec), and chloroacetate esterase staining (Leder staining; Muto) according to the manufacturer’s instructions. The sections were analyzed using a Leica LD2500 microscope at 40× power. Liver and kidney sections were stained with Masson trichrome (Muto) and Prussian blue to visualize necrotic areas and detect iron, respectively. Necrotic areas were measured for each mouse using BZ Analyzer software (KEYENCE) and expressed as the percent of the total area.
Evans blue extravasation analysis
Extravasation of Evans blue was visualized as previously described.23 Mice were anesthetized with a cocktail of fentanyl, midazolam, and acepromazine. The anesthetized mice were injected via the tail vein with 100 μL of Evans blue (Nacalai Tesque), which was allowed to circulate for 1 hour. The back skin was visualized under a microscope (Leica MZ), and photographs of Evans blue leakage were taken. Then, the livers and lungs were harvested and incubated in formamide for 48 hours, and the organs were dried at 56°C for 48 hours and weighed. The extracted Evans blue content was corrected for hemoglobin by measuring the absorbance at 610 and 450 nm. The Evans blue/hemoglobin ratio was calculated and adjusted with the organ dry weight.
Statistical analysis
The bar graphs in all figures represent the mean ± standard deviation. The circles and squares represent single values, and the black bars are error bars. The Mann-Whitney U test was used to calculate statistical significance (P) in Prism 7 software. The log-rank statistic was employed to test whether the survival distributions differed between groups. Statistical significance was indicated by *P < .05 or **P < .01.
Results
Nrf2 activation in monocytes/granulocytes ameliorates organ damage in SCD mice
To determine the beneficial effect of Nrf2 activation in particular cells, we conditionally induced Nrf2 in monocytes/granulocytes by deleting the Keap1 gene, a negative regulator of Nrf2, in SCD mice9,24 by breeding 2 distinct mouse genotypes. Nonphenotypic floxed-Keap1 (referred to as Keap1F/F) mice, which were previously described,21 were inbred with LysM-Cre mice to generate myeloid cell–specific Keap1-deficient mice25 (Keap1F/F::LysM-Cre).
We confirmed the activation of Nrf2 based on the upregulated expression of reduced nicotinamide adenine dinucleotide phosphate:quinone oxidoreductase (Nqo1) messenger RNA (mRNA) in peritoneal macrophages (supplemental Figure 1A) as well as Nqo1 and heme oxygenase 1 (Hmox1) in the livers, lungs, kidneys, and aortas. As expected, Nqo1 mRNA expression was significantly higher in the livers, lungs, kidneys, and aortas of SCD::Keap1F/F::LysM-Cre mice than in those of SCD::Keap1F/F mice, showing increases of approximately twofold, fivefold, more than fourfold, and threefold, respectively (supplemental Figure 1B). Similarly, Hmox1 mRNA expression was much higher in the livers and kidneys of SCD::Keap1F/F::LysM-Cre mice than in those of SCD::Keap1F/F mice (greater than twofold and greater than sixfold higher, respectively), whereas the expression of Hmox1 mRNA was threefold higher in the lungs of SCD::Keap1F/F::LysM-Cre mice than in those of SCD::Keap1F/F mice (supplemental Figure 1C). Our results confirmed the activation of Nrf2 in SCD::Keap1F/F::LysM-Cre mice. We also confirmed the recombination of Keap1 in the lungs, liver, spleen, kidney, and peritoneal macrophages of SCD::Keap1F/F::LysM-Cre mice based on the presence of the 288-bp amplicon from the knockout allele by polymerase chain reaction (supplemental Figure 2A-B). Except for the deletion of Keap1 in SCD::Keap1F/F::LysM-Cre mice, no other significant phenotypic changes were observed. Body weight and organ weight were within the same range in both genotypes (supplemental Figure 2C).
To examine whether Nrf2 activation in myeloid cells affects the RBC phenotype of SCD, we analyzed RBC indices, reticulocyte counts, and RBC lifespan. We found that RBC numbers and hemoglobin levels were moderately but significantly lower in SCD::Keap1F/F::LysM-Cre mice than in SCD::Keap1F/F mice, indicating that anemia was not relieved by Nrf2 activation (supplemental Figure 2D). Reticulocyte counts were comparable between SCD::Keap1F/F and SCD::Keap1F/F::LysM-Cre mice (supplemental Figure 3A). In addition, the lifespan of RBCs was not altered between SCD::Keap1F/F and SCD::Keap1F/F::LysM-Cre mice (supplemental Figure 3B-C). These results indicate that hemolysis is not rescued by Nrf2 activation in myeloid cells.
Genetic alteration of Keap1 can upregulate Nrf2 in SCD mice and improve lung and liver inflammation.12 To assess the effects of Nrf2 in the lungs, we examined the histology of the lungs. Congestion and edema associated with increased myeloid inflammatory cell infiltration were observed in the lungs of SCD::Keap1F/F mice (Figure 1Ai,iii,v), whereas the lung images from SCD::Keap1F/F::LysM-Cre mice showed significant amelioration of these abnormalities (Figure 1Aii,iv,vi). In accordance with the low-grade inflammation observed in SCD::Keap1F/F::LysM-Cre mice, the mRNA expression of the proinflammatory cytokines Tnfα and IL1β, both of which were elevated26 in SCD, was 10 times lower in the lungs of SCD::Keap1F/F::LysM-Cre mice than in those of SCD::Keap1F/F mice (Figure 1B). It has been reported that SCD patients exhibit chronic inflammation, marked by elevated leukocyte counts.27 To verify whether Nrf2 activation in monocytes/granulocytes alters the leukocyte count, we measured the WBC counts of both genotypes. Nrf2 activation in SCD::Keap1F/F::LysM-Cre mice induced a substantial decrease in the WBC count to less than half of that in SCD::Keap1F/F mice (400 × 102/µL vs 800 × 102/μL) (Figure 1C). The proportions of leukocytes in the peripheral blood were comparable between SCD::Keap1F/F and SCD::Keap1F/F::LysM-Cre mice (supplemental Figure 3D). These data clearly show that Nrf2 activation in the monocytes/granulocytes of SCD mice efficiently represses inflammation.
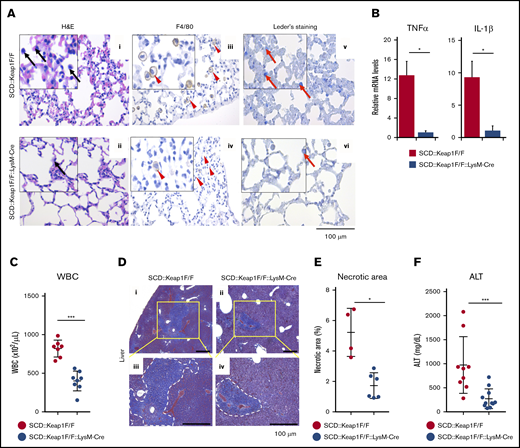
Specific activation of Nrf2 in monocytes/granulocytes alleviates inflammatory damage. (A) hematoxylin and eosin (H&E), F4/80, and chloroacetate esterase (Leder staining) staining of lungs from SCD::Keap1F/F (i,iii,v) mice and SCD::Keap1F/F::LysM-Cre (ii,iv,vi) mice showing congestion of the lungs and infiltration of inflammatory cells (i-ii), specifically macrophages (iii-iv) and neutrophils (v-vi). Black arrows show myeloid cells; arrowheads indicate macrophages, and red arrows denote neutrophils. Scale bar, 100 µm. (B) mRNA expression levels of tumor necrosis factor-α (TNF-α; left) and interleukin-1β (IL-1β; right) in the lungs of SCD::Keap1F/F (n = 6) and SCD::Keap1F/F::LysM-Cre mice (n = 6). (C) WBC counts in the peripheral blood of SCD::Keap1F/F (n = 7) and SCD::Keap1 F/F::LysM-Cre mice (n = 8). (D) Masson trichrome staining of livers from SCD::Keap1F/F (i,iii) and SCD::Keap1 F/F::LysM-Cre mice (ii,iv). Necrotic areas are delimited by dotted white lines. The areas in yellow squares are enlarged in panels iii-iv. (E) Quantification of necrotic areas in the liver. (F) Liver ALT levels of SCD::Keap1F/F (n = 9) and SCD::Keap1F/F::LysM-Cre mice (n = 11). The bar graphs represent the mean ± standard deviation. *P < .05; ***P < .001.
Specific activation of Nrf2 in monocytes/granulocytes alleviates inflammatory damage. (A) hematoxylin and eosin (H&E), F4/80, and chloroacetate esterase (Leder staining) staining of lungs from SCD::Keap1F/F (i,iii,v) mice and SCD::Keap1F/F::LysM-Cre (ii,iv,vi) mice showing congestion of the lungs and infiltration of inflammatory cells (i-ii), specifically macrophages (iii-iv) and neutrophils (v-vi). Black arrows show myeloid cells; arrowheads indicate macrophages, and red arrows denote neutrophils. Scale bar, 100 µm. (B) mRNA expression levels of tumor necrosis factor-α (TNF-α; left) and interleukin-1β (IL-1β; right) in the lungs of SCD::Keap1F/F (n = 6) and SCD::Keap1F/F::LysM-Cre mice (n = 6). (C) WBC counts in the peripheral blood of SCD::Keap1F/F (n = 7) and SCD::Keap1 F/F::LysM-Cre mice (n = 8). (D) Masson trichrome staining of livers from SCD::Keap1F/F (i,iii) and SCD::Keap1 F/F::LysM-Cre mice (ii,iv). Necrotic areas are delimited by dotted white lines. The areas in yellow squares are enlarged in panels iii-iv. (E) Quantification of necrotic areas in the liver. (F) Liver ALT levels of SCD::Keap1F/F (n = 9) and SCD::Keap1F/F::LysM-Cre mice (n = 11). The bar graphs represent the mean ± standard deviation. *P < .05; ***P < .001.
Organ infarcts are a hallmark of SCD pathology, and the liver is one of the most affected organs. We evaluated liver morphology, and the histological examination revealed that liver sections from SCD::Keap1F/F mice were characterized by abundant focal areas of necrosis and early inflammatory regions (Figure 1Di,iii), whereas liver sections from SCD::Keap1 F/F::LysM-Cre mice exhibited much smaller necrotic areas (Figure 1Dii,iv) than those of SCD::Keap1F/F mice. Upon measuring the areas of necrosis in these mice, we found a significant difference (Figure 1E). ALT is a marker of liver damage. Although the mean plasma ALT value was still higher than that of normal mice, the mean ALT value of SCD::Keap1F/F::LysM-Cre mice was >2 times lower than that of SCD::Keap1F/F mice (341 mg/dL vs 641 mg/dL) (Figure 1F). These results are in agreement with the data obtained under systemic Nrf2 activation in SCD mice12 and indicate that Nrf2 activation in monocytes/granulocytes explains a major part of the improvement in SCD.
SCD::Keap1F/F::LysM-Cre mice exhibit mitigated heme accumulation
Liver Kupffer cells, spleen red-pulp macrophages, and, to some extent, bone marrow macrophages phagocytose aged and/or damaged RBCs; through this process, heme is degraded into less toxic molecules and iron.28 To study the effect of Nrf2 activation in macrophages on heme metabolism, we performed an imaging mass spectrometry (iMScope; Shimadzu, Kyoto, Japan) analysis to visualize the distribution of heme (m/z 616) in the liver and spleen. Although the heme content in the spleens of both SCD::Keap1F/F and SCD::Keap1F/F::LysM-Cre mice was considerably increased, the mean content of heme was slightly but not significantly decreased in SCD::Keap1F/F::LysM-Cre spleens (supplemental Figure 4). In contrast, the livers of SCD::Keap1F/F::LysM-Cre mice showed significantly lower heme signals in the parenchyma and vessels, whereas those of SCD::Keap1F/F mice displayed strong signals in their vessels and parenchyma (Figure 2A-B). This result represents the first direct demonstration by bioimaging that the heme distribution in the livers of SCD mice is modulated by Nrf2 activation.
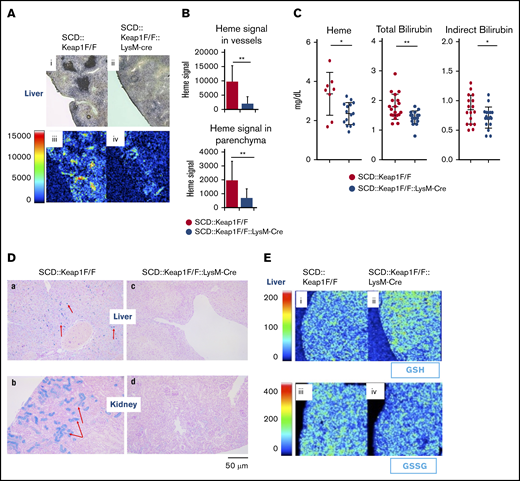
Keap1 deletion in monocytes/granulocytes improves heme and iron metabolism. (A) iMScope analysis of heme in SCD::Keap1F/F (i,iii) and SCD::Keap1F/F::LysM-Cre mouse livers (ii,iv). Bright-field images are shown in the upper panel (i-ii), and the yellow and red regions in the lower panel (iii-iv) represent heme accumulation in liver vessels and parenchyma. (B) Quantification of heme ion emissions in the vessels of the liver (upper) and parenchyma (lower) of SCD::Keap1F/F mice and SCD::Keap1F/F::LysM-Cre mice. (C) Plasma heme levels (left), plasma total bilirubin levels (middle), and plasma indirect bilirubin levels (right) of SCD::Keap1F/F mice and SCD::Keap1F/F LysM-Cre mice. (D) Prussian blue staining of iron in the livers (a,c) and kidneys (b,d) of SCD::Keap1F/F mice (a-b) and SCD::Keap1F/F::LysM-Cre mice (c-d). The red arrows show iron accumulation. Scale bar, 50 μm. (E) Matrix-assisted laser desorption/ionization iMScope of GSH (upper; i-ii) and GSSG (lower; iii-iv) in livers from SCD::Keap1F/F mice and SCD::Keap1F/F::LysM-Cre mice via iMScope. *P < .05; **P < .01.
Keap1 deletion in monocytes/granulocytes improves heme and iron metabolism. (A) iMScope analysis of heme in SCD::Keap1F/F (i,iii) and SCD::Keap1F/F::LysM-Cre mouse livers (ii,iv). Bright-field images are shown in the upper panel (i-ii), and the yellow and red regions in the lower panel (iii-iv) represent heme accumulation in liver vessels and parenchyma. (B) Quantification of heme ion emissions in the vessels of the liver (upper) and parenchyma (lower) of SCD::Keap1F/F mice and SCD::Keap1F/F::LysM-Cre mice. (C) Plasma heme levels (left), plasma total bilirubin levels (middle), and plasma indirect bilirubin levels (right) of SCD::Keap1F/F mice and SCD::Keap1F/F LysM-Cre mice. (D) Prussian blue staining of iron in the livers (a,c) and kidneys (b,d) of SCD::Keap1F/F mice (a-b) and SCD::Keap1F/F::LysM-Cre mice (c-d). The red arrows show iron accumulation. Scale bar, 50 μm. (E) Matrix-assisted laser desorption/ionization iMScope of GSH (upper; i-ii) and GSSG (lower; iii-iv) in livers from SCD::Keap1F/F mice and SCD::Keap1F/F::LysM-Cre mice via iMScope. *P < .05; **P < .01.
Because SCD::Keap1F/F::LysM-Cre mice showed a low heme distribution in the liver, we speculated that this could be a result of long-term heme accumulation and that measuring plasma heme levels could provide insight into the actual hemolysis status. Therefore, we measured plasma heme and downstream metabolites. The assessment of plasma heme revealed that SCD::Keap1F/F::LysM-Cre mice exhibited substantially lower levels of circulating free heme, whereas compared with SCD::Keap1F/F mice, SCD::Keap1F/F::LysM-Cre mice showed altered plasma levels of both total bilirubin and indirect bilirubin (Figure 2C). These results suggest that Nrf2 activation in myeloid cells is critical for enhancing the cellular metabolism of heme and reducing circulating free heme.
We further evaluated the iron content and glutathione (GSH) distribution in the liver and kidneys. Prussian blue staining revealed a ubiquitous iron distribution in the livers of SCD::Keap1F/F mice (Figure 2Da), whereas iron accumulation was specifically confined to the collecting ducts in the kidneys of SCD::Keap1F/F mice (Figure 2Db). In contrast, iron accumulation was practically nonexistent in the livers and kidneys of SCD::Keap1F/F::LysM-Cre mice (Figure 2Dc-d).
Based on a previous report on GSH-mediated attenuation of heme-induced adhesion molecule expression in ECs,29 we surmised that the GSH level may be upregulated in SCD::Keap1F/F::LysM-Cre mice. To address this hypothesis, we examined the GSH distribution with iMScope and found that the signal intensity of GSH (m/z 306.07) was significantly stronger in SCD::Keap1F/F::LysM-Cre mice than in SCD::Keap1F/F mice (Figure 2Ei-ii). This observation is in agreement with our hypothesis that Nrf2 acts to enhance the antioxidant activity of cells. In contrast, the signal of the oxidized form of GSH, glutathione disulfide (GSSG), was weaker in SCD::Keap1F/F::LysM-Cre mouse livers than in SCD::Keap1F/F mouse livers (Figure 2Eiii,iv). These results demonstrate that Nrf2 activation in SCD mice results in increases in GSH and decreases in the oxidized form of GSH (GSSG) in the liver.
Nrf2 activation in ECs ameliorates organ damage in SCD mice
To assess whether Nrf2 activation in ECs contributes to the amelioration of the SCD phenotype, we generated mice in which Nrf2 was activated specifically in ECs by crossing SCD::Keap1F/F mice with Tie1-Cre mice (SCD::Keap1F/F::Tie1-Cre mice). We confirmed the recombination of Keap1 in the lungs, livers, spleens, kidneys, and mouse primary pulmonary endothelial cells (MPPECs) of SCD::Keap1F/F::Tie1-Cre mice based on the presence of the 288-bp amplicon from the knockout allele by polymerase chain reaction (supplemental Figure 5A-B). In addition, we confirmed that Keap1 deletion and Nrf2 activation did not occur in these mice (supplemental Figure 5C). We noted significant decreases in the RBC count and hemoglobin in SCD::Keap1F/F::Tie1-Cre mice (supplemental Figure 5D). There were no significant differences in reticulocyte counts or RBC lifespan between SCD::Keap1F/F and SCD::Keap1F/F::Tie-Cre mice (supplemental Figure 3A-C). These results indicate that anemia and hemolysis are not improved by Nrf2 activation in ECs. We examined WBC counts in both SCD::Keap1F/F and SCD::Keap1F/F::Tie1-Cre mice and found that EC-specific Nrf2 activation reduced the number of WBCs by twofold in SCD::Keap1F/F::Tie1-Cre mice compared with that in SCD::Keap1F/F mice (Figure 3A). Except for a slight increase in myeloid cells, the proportions of leukocytes in the peripheral blood were comparable between SCD::Keap1F/F and SCD::Keap1F/F::Tie-Cre mice (supplemental Figure 3D). Importantly, the magnitude of this decrease was similar to that observed in SCD::Keap1F/F::LysM-Cre mice.
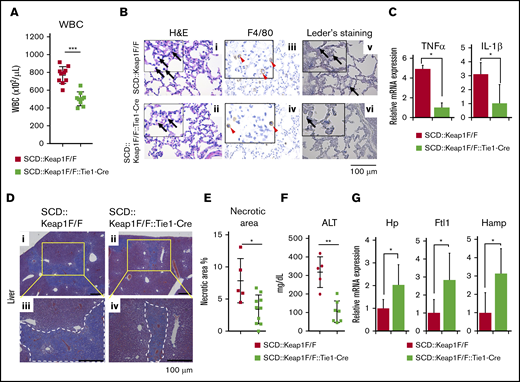
EC-specific deletion of Keap1 rescues inflammation and liver function. (A) WBC counts in the peripheral blood of SCD::Keap1F/F mice (n = 10) and SCD::Keap1F/F::Tie1-Cre mice (n = 9). (B) H&E, F4/80, and chloroacetate esterase (Leder staining) staining of lungs from SCD::Keap1F/F (i,iii,v) mice and SCD::Keap1F/F::Tie1-Cre (ii,iv,vi) mice showing infiltration of macrophages (iii-iv) and neutrophils (v-vi) and congestion of the lungs of SCD::Keap1 F/F mice. Black arrows show myeloid cells and neutrophils; arrowheads indicate macrophages, and red arrows denote neutrophils. (C) Quantification of the mRNA levels of TNF-α (upper) and IL-1β (lower) in the lungs of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 6). (D) Masson trichrome staining of livers from SCD::Keap1F/F mice (i,iii) and SCD::Keap1F/F::Tie1-Cre mice (ii,iv). Scale bar, 100 µm. (E) Quantification analysis of necrotic areas in the liver. Necrotic areas were measured in each mouse using BZ Analyzer Software (KEYENCE) and are expressed as the percent of the total area. (F) Liver ALT levels of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 7). (G) Quantification of the mRNA levels of Hp, Ftl1, and Hamp in the livers of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 5). The bar graphs represent the mean ± standard deviation. *P < .05; **P < .01; ***P < .001.
EC-specific deletion of Keap1 rescues inflammation and liver function. (A) WBC counts in the peripheral blood of SCD::Keap1F/F mice (n = 10) and SCD::Keap1F/F::Tie1-Cre mice (n = 9). (B) H&E, F4/80, and chloroacetate esterase (Leder staining) staining of lungs from SCD::Keap1F/F (i,iii,v) mice and SCD::Keap1F/F::Tie1-Cre (ii,iv,vi) mice showing infiltration of macrophages (iii-iv) and neutrophils (v-vi) and congestion of the lungs of SCD::Keap1 F/F mice. Black arrows show myeloid cells and neutrophils; arrowheads indicate macrophages, and red arrows denote neutrophils. (C) Quantification of the mRNA levels of TNF-α (upper) and IL-1β (lower) in the lungs of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 6). (D) Masson trichrome staining of livers from SCD::Keap1F/F mice (i,iii) and SCD::Keap1F/F::Tie1-Cre mice (ii,iv). Scale bar, 100 µm. (E) Quantification analysis of necrotic areas in the liver. Necrotic areas were measured in each mouse using BZ Analyzer Software (KEYENCE) and are expressed as the percent of the total area. (F) Liver ALT levels of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 7). (G) Quantification of the mRNA levels of Hp, Ftl1, and Hamp in the livers of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 5). The bar graphs represent the mean ± standard deviation. *P < .05; **P < .01; ***P < .001.
Furthermore, inflammatory cell infiltration and congestion in the lungs were significantly lower in SCD::Keap1F/F::Tie1-Cre mice than in SCD::Keap1F/F mice (Figure 3B). Consistent with the low levels of inflammatory cells in the blood and lungs, EC-specific Nrf2 activation appeared to robustly repress the mRNA expression of proinflammatory cytokines, notably TNF-α and IL-1β (Figure 3C).
Because Nrf2 activation in myeloid cells (ie, SCD::Keap1F/F::LysM-Cre) was shown to be critical for liver protection, we next examined whether Nrf2 activation in ECs exerted similar effects in SCD::Keap1F/F::Tie1-Cre mice. Notably, necrotic lesions caused by acute and chronic liver inflammation were significantly lower in SCD::Keap1F/F::Tie1-Cre mice (Figure 3Dii,iv) than in SCD Keap1 F/F mice (Figure 3Di,iii). The necrotic areas were decreased by almost half upon EC-specific Nrf2 activation (Figure 3E). Although the livers of SCD::Keap1F/F::Tie1-Cre mice exhibited less necrosis, iMScope analysis failed to demonstrate a significant change in heme accumulation (supplemental Figure 6). We also measured plasma ALT levels and found that the level in SCD::Keap1F/F::Tie1-Cre mice was decreased by more than half (Figure 3F).
Heme and iron-scavenging proteins are critical for maintaining iron homeostasis and regulating the pool of free heme, consequently limiting the amount of heme available as a catalyst of free radical formation. A previous report showed that administration of the Nrf2 activator dimethyl fumarate ameliorates liver damage and induces haptoglobin (Hp).30 Because liver damage was relieved in SCD::Keap1F/F::Tie1-Cre mice, we measured the mRNA expression levels of Hp, Ftl1, and hepcidin (Hamp) in the liver. All of these genes were upregulated by two- to threefold in SCD::Keap1F/F::Tie1-Cre mice compared with SCD::Keap1F/F mice (Figure 3G). Taken together, these results demonstrate that Nrf2 mediates the resolution of inflammation and tissue protection in SCD mice by activating the cytoprotective ability of ECs.
Nrf2 activation protects the vascular endothelium
Vascular pathology is a key component of inflammation, vasoocclusion, and heme extravasation in SCD.31 Because ECs are on the frontline of hemolytic aggression, we hypothesized that Nrf2 might protect vascular function from oxidative insults. Our analysis of the mRNA expression of vasoocclusion mediators, such as vascular cell adhesion molecule 1 (Vcam1) and P-selectin, in the lungs showed lower mRNA expression of these molecules in SCD::Keap1F/F::Tie1-Cre mice than in SCD::Keap1F/F mice (Figure 4A). This result suggests that the adhesion of inflammatory cells is attenuated by the activation of Nrf2 in ECs.
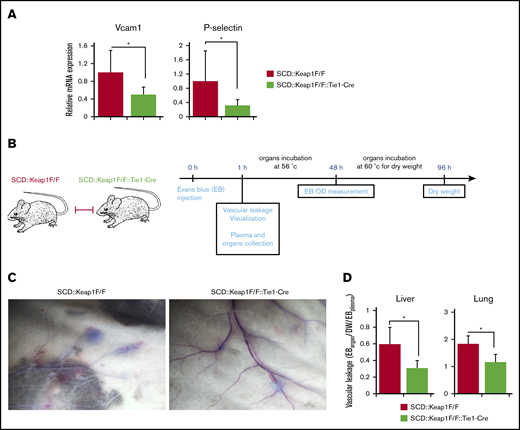
Nrf2 activation in the ECs of SCD mice alters vascular leakage in vivo. (A) Quantification of the mRNA levels of Vcam1 (left) and P-selectin (right) in the lungs of SCD::Keap1F/F mice (n = 8) and SCD::Keap1F/F::Tie1-Cre mice (n = 8). (B) Schematic illustration of the Evans blue (EB) experiment. (C) Nrf2 activation in SCD::Keap1F/F::Tie1-Cre mice abrogates vascular leakage in the back skin. (D) Quantification of the ratio of formamide-extracted EB by spectrophotometry (610 nm), corrected for dry weight (DW) and hemoglobin (450 nm), in the livers and lungs of SCD::Keap1F/F and SCD::Keap1F/F::Tie1-Cre mice. *P < .05 (n = 5). OD, optical density.
Nrf2 activation in the ECs of SCD mice alters vascular leakage in vivo. (A) Quantification of the mRNA levels of Vcam1 (left) and P-selectin (right) in the lungs of SCD::Keap1F/F mice (n = 8) and SCD::Keap1F/F::Tie1-Cre mice (n = 8). (B) Schematic illustration of the Evans blue (EB) experiment. (C) Nrf2 activation in SCD::Keap1F/F::Tie1-Cre mice abrogates vascular leakage in the back skin. (D) Quantification of the ratio of formamide-extracted EB by spectrophotometry (610 nm), corrected for dry weight (DW) and hemoglobin (450 nm), in the livers and lungs of SCD::Keap1F/F and SCD::Keap1F/F::Tie1-Cre mice. *P < .05 (n = 5). OD, optical density.
To analyze vascular endothelium function, we IV administered Evans blue solution to mice and visualized its extravasation 1 hour after injection (Figure 4B). Images of the dorsal skin of SCD::Keap1F/F mice displayed a large amount of Evans blue leakage from narrow vessels; in contrast, the amount of Evans blue leakage was attenuated in SCD::Keap1F/F::Tie1-Cre mice (Figure 4C). We also evaluated the integrity of vessels in the livers and lungs. Following the injection of Evans blue, lungs and livers were isolated in formamide and processed as previously described.32 Compared with SCD::Keap1F/F::Tie1-Cre mice, SCD::Keap1F/F mice showed a twofold increase in Evans blue extracted from both the liver and the lungs (Figure 4D). These results support our hypothesis that Nrf2 controls vascular occlusion as well as vascular integrity by regulating EC cohesion and that Nrf2 prevents vascular pathology in SCD.
MPPECs express Nrf2-dependent genes
To identify the molecular basis of the benefits of Nrf2 to ECs in SCD mice, we next analyzed the gene expression profiles of MPPECs from 7- to 12-day-old SCD::Keap1F/F mice and SCD::Keap1F/F::Tie1-Cre mice through RNA-sequencing analysis (Figure 5A). The mRNA expression of Nqo1 was threefold higher in SCD::Keap1F/F::Tie-Cre cells (Figure 5B). RNA-sequencing analysis revealed that 879 genes were significantly upregulated in SCD::Keap1F/F::Tie-Cre cells compared with SCD::Keap1F/F cells, and 898 genes were significantly downregulated in SCD::Keap1F/F cells compared with SCD::Keap1 F/F::Tie-Cre cells (Figure 5C).
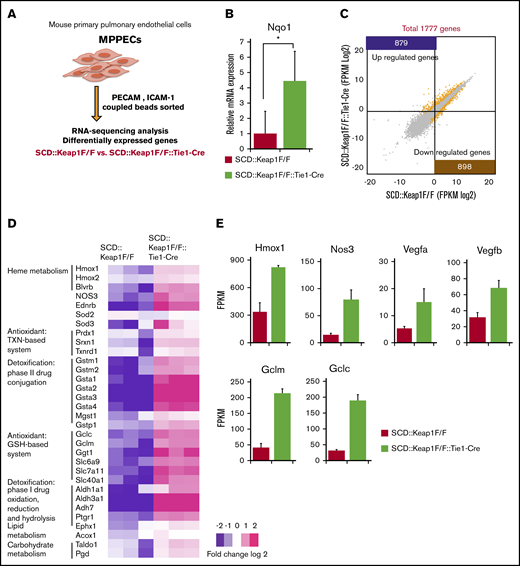
Keap1 deletion in ECs upregulates the cell defense mechanism related to Nrf2. (A) Model of MPPEC culture and analysis. (B) Relative mRNA levels of Nqo1 in the MPPECs of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 5). *P < .05. (C) Scatter plots comparing transcript levels in SCD::Keap1F/F (x-axis) and SCD::Keap1F/F::Tie1-Cre (y-axis) MPPECs. A total of 879 (purple) genes were significantly upregulated, and 898 (mustard) genes were significantly downregulated in SCD Keap1F/F::Tie1-Cre MPPECs compared with those in SCD::Keap1F/F MPPECs. (D) Heat map showing relative expression levels in SCD::Keap1F/F (left) and SCD::Keap1F/F::Tie1-Cre mice (right). (E) Expression levels of Hmox1, Nos3, Vegfa, Vegfb, Gclm, and Gclc as fragments per kilobyte of exons per million fragments (FPKM) read values. TXN, thioredoxin.
Keap1 deletion in ECs upregulates the cell defense mechanism related to Nrf2. (A) Model of MPPEC culture and analysis. (B) Relative mRNA levels of Nqo1 in the MPPECs of SCD::Keap1F/F mice (n = 5) and SCD::Keap1F/F::Tie1-Cre mice (n = 5). *P < .05. (C) Scatter plots comparing transcript levels in SCD::Keap1F/F (x-axis) and SCD::Keap1F/F::Tie1-Cre (y-axis) MPPECs. A total of 879 (purple) genes were significantly upregulated, and 898 (mustard) genes were significantly downregulated in SCD Keap1F/F::Tie1-Cre MPPECs compared with those in SCD::Keap1F/F MPPECs. (D) Heat map showing relative expression levels in SCD::Keap1F/F (left) and SCD::Keap1F/F::Tie1-Cre mice (right). (E) Expression levels of Hmox1, Nos3, Vegfa, Vegfb, Gclm, and Gclc as fragments per kilobyte of exons per million fragments (FPKM) read values. TXN, thioredoxin.
Recurrent vasoocclusion in SCD seems to facilitate ischemia-reperfusion injury with subsequent ROS production. Moreover, hemolysis triggers oxidation and peroxidation reactions in various cells, facilitating the activation of important cells in vascular pathology, such as leukocytes, neutrophils, macrophages, and ECs. These events are associated with further ROS production, which at a certain point overwhelms the endogenous mechanism of detoxification. Our RNA-sequencing results showed modification of known Nrf2 target pathways in SCD::Keap1F/F::Tie1-Cre cells; among these pathways, heme metabolism and the antioxidant thioredoxin and GSH-based system were both significantly upregulated (Figure 5D). Among the upregulated pathways, we identified direct Nrf2 target genes such as Hmox1, nitric oxide synthase 3 (Nos3), vascular endothelial growth factor a (Vegfa), and vascular endothelial growth factor b (Vegfb), which are responsible for the heme degradation and vascular relaxation critical to SCD, as well as the glutamate-cysteine ligase modifier subunit (Gclm) and glutamate-cysteine ligase catalytic subunit (Gclc) (Figure 5E). Moreover, other pathways, such as IkB kinase NF-KappaB signaling regulation, ion homeostasis, and PI3K-Akt signaling, were enriched in SCD::Keap1F/F::Tie1-Cre ECs (supplemental Figure 7A-B). Thus, these results indicate that Nrf2 activation in ECs contributes, to some extent, to attenuating the burst of oxidative molecule production following hemolysis and ischemia-reperfusion injury and protecting vascular function.
Discussion
Until recently, hydroxyurea was the only medication approved to treat SCD, but even in cases in which hydroxyurea shows good efficacy, patients can still develop organ dysfunction and a range of comorbid complications of SCD. In 2017, the Food and Drug Administration approved l-glutamine (Endari) as a new medication for patients aged 5 years and older with SCD.33 Because there are still concerns about the therapeutic response and comorbid complications of these treatments,34-36 the development of new treatments for SCD is awaited. In this regard, drugs based on epigenetic regulation/activation of the γ-globin gene have been scrutinized for a decade37 as alternatives or complementary treatments to hydroxyurea. Through a distinct approach, we recently demonstrated that Nrf2 activation via genetic alteration or chemical repression of its repressor Keap1 significantly suppresses ischemia-reperfusion injury. Nrf2 ameliorates inflammation-mediated damage to tissues and preserves organ function in SCD mice.12 The present study extended those results and investigated the cell type–specific roles that Nrf2 plays in the protection of organs/tissues from SCD pathology. We provide solid in vivo lines of evidence that Nrf2 activation in myeloid lineage cells and ECs contributes cooperatively to the alleviation of hemolysis-related inflammation and ischemia-reperfusion organ injury in SCD model mice.
The targeted Keap1 gene deletion exploited in this study was expected to upregulate Nrf2 and its downstream enzymes specifically in monocytes/granulocytes and ECs. In these models, flox modification of the Keap1 gene did not induce any deleterious phenotype in SCD::Keap1F/F mice because the Keap1 gene remained intact in the mice. In SCD::Keap1F/F::LysM-Cre and SCD::Keap1F/F::Tie1-Cre mice, deletion of exons 2 and 3 of the Keap1 gene resulted in loss of Keap1 activity.21 As shown in Figure 6, the overexpression of Nrf2 in ECs upregulated the expression of Hmox1 and other critical enzymes necessary to neutralize ROS generated through hemolysis, thereby protecting ECs against inflammatory activation. Consequently, the interactions of ECs with other inflammatory cells were disrupted, as demonstrated by the low expression levels of Vcam1 and P-selectin. Furthermore, Nrf2 activation improved vascular integrity by counteracting vascular leakage and thereby preventing edema formation in the extracellular compartment. Following Nrf2 activation, macrophages overexpressed heme metabolism enzymes such as HO-1 (encoded by Hmox1) and biliverdin reductase, and elimination of the toxic forms of iron and heme was enhanced. As a result, oxidative stress was enfeebled, and the expression of proinflammatory cytokines was reduced, partly through the downregulation of heme-mediated cell activation. Overall, the activation of Nrf2 in both cell lineages (ie, monocytes/granulocytes and ECs) conferred strong and efficient amelioration of the SCD phenotypes.
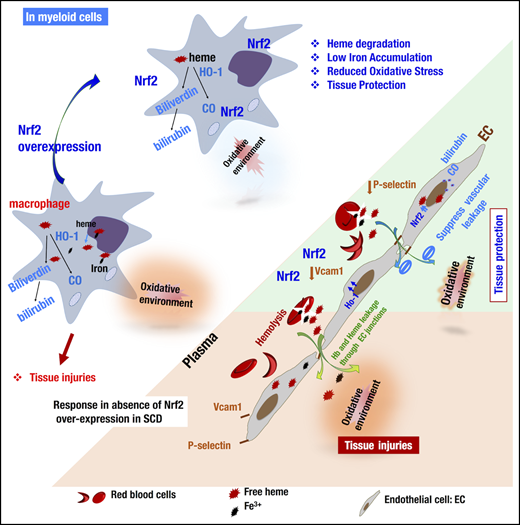
Model illustrating how Nrf2 activation via the deletion of Keap1 in ECs or monocytes/granulocytes contributes individually to ameliorating the SCD phenotype. In SCD, chronic hemolysis alters the balance between the production and detoxification of antioxidants. In macrophages, for instance (left), Nrf2 insufficiency impairs the safe storage and metabolism of iron ions and free heme. Upon Nrf2 overexpression, macrophages can break down and store free heme and iron in less toxic molecules and decrease the production of oxidative species. In ECs (right), hemolysis and lower levels of Nrf2 promote the activation of ECs, expression of adhesion molecules, and impairment of endothelial barrier integrity (orange board). Activation of Nrf2 (green panel) inhibits the expression of Vcam1 and P-selectin, upregulates the expression of antioxidant enzymes, and, last but not least, inhibits vascular leakage by restoring the integrity of the vascular endothelium. EC-specific or monocyte/granulocyte-specific activation of Nrf2 ultimately protects organs from inflammation and damage in a distinct manner.
Model illustrating how Nrf2 activation via the deletion of Keap1 in ECs or monocytes/granulocytes contributes individually to ameliorating the SCD phenotype. In SCD, chronic hemolysis alters the balance between the production and detoxification of antioxidants. In macrophages, for instance (left), Nrf2 insufficiency impairs the safe storage and metabolism of iron ions and free heme. Upon Nrf2 overexpression, macrophages can break down and store free heme and iron in less toxic molecules and decrease the production of oxidative species. In ECs (right), hemolysis and lower levels of Nrf2 promote the activation of ECs, expression of adhesion molecules, and impairment of endothelial barrier integrity (orange board). Activation of Nrf2 (green panel) inhibits the expression of Vcam1 and P-selectin, upregulates the expression of antioxidant enzymes, and, last but not least, inhibits vascular leakage by restoring the integrity of the vascular endothelium. EC-specific or monocyte/granulocyte-specific activation of Nrf2 ultimately protects organs from inflammation and damage in a distinct manner.
Endogenous mechanisms required for the phagocytosis of damaged RBCs and plasma heme/hemoglobin clearance in macrophages can be easily overloaded in SCD patients suffering from chronic hemolysis. Heme and iron can induce a phenotypic switch of macrophages toward proinflammatory activity and compromise their erythrophagocytic function.38 RBC sickling and hemolysis are characteristics of the SCD phenotype; although Nrf2 activation is insufficient to reverse the sickling of RBCs,12 the deleterious effects of hemolysis have been shown to be altered. It has been reported that transcription of the IL-1β and IL-6 genes is negatively regulated in macrophages overexpressing Nrf2.8 Because hemolysis can trigger an inflammatory response and elicit cell damage, we surmise that the activation of Nrf2 in monocytes/granulocytes impairs the SCD phenotype by critically upregulating antioxidant mechanisms and inhibiting factors that trigger inflammation.
Although the activation of Nrf2 in monocytes/granulocytes appears to give rise to similar consequences to that in ECs, we believe that the mechanisms underlying the 2 processes are distinct but cooperative. In monocytes/granulocytes and ECs, Nrf2 upregulates defense mechanisms against ROS. However, SCD::Keap1F/F::LysM-Cre mice show a greater increase in heme metabolism and reduction in heme deposits in tissues than SCD::Keap1F/F::Tie1-Cre mice. Moreover, in SCD::Keap1F/F::Tie-Cre mice, Keap1 disruption leads to vascular leakage blockage by preserving the function of the endothelium and upregulating the expression of heme and iron scavengers. In our study, targeted activation of Nrf2 in either monocytes/granulocytes or ECs appeared to modify the entire SCD phenotype. In SCD mice, we surmise that this specific-cell activation of Nrf2 has a positive or negative influence on neighboring cells that in turn gives rise to beneficial effects for tissue protection. Thus, we may explain how Nrf2 activation in ECs modifies the expression of Hp and Hamp in the liver.
We found significant decreases in the RBC and hemoglobin concentration upon Nrf2 activation in ECs (supplemental Figure 5D); however, there were no significant differences in reticulocyte counts (production) or RBC lifespan (degradation) (supplemental Figure 3A-C). On the other hand, we observed that plasma Evans blue leakage was attenuated by Nrf2 activation in ECs (Figure 4C). Based on these results, we surmise that the volume of plasma was higher in SCD::Keap1F/F::Tie1-Cre mice than in SCD::Keap1F/F mice, and the RBC and hemoglobin were thus relatively decreased.
Nrf2 may also contribute to the protection of erythroid lineage cells in SCD. In fact, the application of Nrf2 inducers increases γ-globin activity and fetal hemoglobin, which are associated with the alleviation of liver tissue inflammation and injury.39,40 In addition, Nrf2 plays a role in preventing oxidative stress in erythrocytes.41 These observations provide alternative lines of evidence that we present in this report. We show in this study that when Nrf2 is activated specifically in the myeloid cells and ECs of SCD model mice, inflammation and tissue damage are ameliorated without anemia, and hemolysis improvement as Nrf2 is not induced in erythroid cells in this case. Consistent with our observations, the contribution of Nrf2 in nonhematopoietic cells has been shown in a study involving bone marrow transplantation experiments.42 We conclude based on these results that Nrf2 activation in erythroid lineage cells, as well as myeloid cells and ECs, contributes to the amelioration of SCD.
Various approaches for SCD treatment, including gene therapy and bone marrow transplantation, have been discussed or assessed in clinical trials.43-45 Drugs that target reactivation of the γ-globin gene are designed to inhibit the polymerization of hemoglobin tetramers and hinder the sickling of RBCs.46-48 Based on the evidence obtained in this study, we propose the combinatory use of Nrf2 inducers with γ-globin inducers for treating SCD. In this regard, loss of Nrf2 was also recently reported to decrease γ-globin expression.49 One Nrf2 activator, Tecfidera, has been used worldwide for the treatment of multiple sclerosis. Bardoxolone is in a phase 3 clinical trial in Japan. Although a phase 3 study of bardoxolone in type 2 diabetes mellitus nephropathy (BEACON) was stopped because of a high rate of cardiovascular events, post hoc analyses showed that the cardiovascular events (fluid overload) were associated with predictable risk factors, such as high brain natriuretic peptide levels and prior history of heart failure.50,51 A Japanese company has conducted phase 2 (TSUBAKI; #NCT02316821) and 3 (AYAME; #NCT03550443) studies by excluding at-risk patients and showed that bardoxolone improved renal function with no signs or symptoms of fluid overload. Therefore, Nrf2 activators are effective in the regulation of risk factors.
To the best of our knowledge, this study is the first to demonstrate specific contributions of Nrf2 activation in myeloid lineage cells and ECs of SCD mice in vivo. In support of this notion, a limited number of studies have investigated Nrf2 function in ECs and macrophages/monocytes using culture cell lines.42,52,53 Vascular pathology is a key component of inflammation, vasoocclusion, and heme extravasation in SCD, and understanding the function of Nrf2 in MPPECs is crucial for the development of effective therapies. On the basis of these mechanistic insights, we surmise that Nrf2 inducers are promising drugs for use either alone or in combination for the treatment of SCD patients and other hemolytic diseases, such as thalassemia.54 The development of in vivo models that target cells other than macrophages and ECs would further contribute to our understanding of SCD pathology.
Acknowledgments
The authors thank Eriko Naganuma and the Biomedical Research Core of Tohoku University Graduate School of Medicine for technical support.
This work was supported in part by KAKENHI grants 15H02507 (M.Y.), 24799957 (M.S.), and 16H06639 (N.K.-L.) from the Japan Society for the Promotion of Science. The authors also acknowledge AMED-P-CREATE (M.Y.) and the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Ministry of Education, Culture, Sports, Science and Technology, and AMED (M.Y.).
Authorship
Contribution: N.K.-L. and M.S. designed the research, performed the experiments, analyzed the data, and wrote the manuscript; H.P. performed the experiments; F.K., A.O., and N.K.-L. organized the RNA-sequencing analysis and performed the bioinformatics analyses; D.S., A.U., and R.S. supervised the IMS experiments; and M.Y. supervised the project and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Masayuki Yamamoto, Department of Medical Biochemistry, Tohoku University Graduate School of Medicine, Sendai 980-8575, Japan; e-mail: masiyamamoto@med.tohoku.ac.jp; and Nadine Keleku-Lukwete, Department of Medical Biochemistry, Tohoku University Graduate School of Medicine, Sendai 980-8575, Japan; e-mail: nadine@med.tohoku.ac.jp.

