

Key Points
After loss of wild-type TP53 expression, the RAS pathway is the second most frequently altered pathway in the CK AML subgroup.
Adverse primary human AML is sensitive to PLK1 inhibitors in vitro and in xenotransplantation mouse models.

Abstract
Patients diagnosed with acute myeloid leukemia with complex karyotype (CK AML) have an adverse prognosis using current therapies, especially when accompanied by TP53 alterations. We hereby report the RNA-sequencing analysis of the 68 CK AML samples included in the Leucegene 415 patient cohort. We confirm the frequent occurrence of TP53 alterations in this subgroup and further characterize the allele expression profile and transcript alterations of this gene. We also document that the RAS pathway (N/KRAS, NF1, PTPN11, BRAF) is frequently altered in this disease. Targeted chemical interrogation of genetically characterized primary CK AML samples identifies polo-like kinase 1 (PLK1) inhibitors as the most selective agents for this disease subgroup. TP53 status did not alter sensitivity to PLK1 inhibitors. Interestingly, CK AML specimens display a G2/M transcriptomic signature that includes higher expression levels of PLK1 and correlates with PLK1 inhibition sensitivity. Together, our results highlight vulnerability in CK AML. In line with these in vitro data, volasertib shows a strong anti-AML activity in xenotransplantation mouse models of human adverse AML. Considering that PLK1 inhibitors are currently being investigated clinically in AML and myelodysplastic syndromes, our results provide a new rationale for PLK1-directed therapy in patients with adverse cytogenetic AML.
Introduction
Complex karyotype acute myeloid leukemia (CK AML) is defined as having 3 or more chromosomal abnormalities1 in the absence of one of the World Health Organization–designated recurrent genetic abnormalities.2 The presence of a CK classifies patients into the adverse risk group, with an expected long-term overall survival <20%.1,3 CK AML forms a genetically heterogeneous subgroup that almost never cooccurs with NPM1 nor with biallelic CEBPA mutations and infrequently carries mutations in FLT3, IDH1, IDH2, DNMT3A, and TET2.4 In sharp contrast, this subgroup is characterized by chromosomal aneuploidies and by structural alterations and/or point mutations in TP53, the latter being observed in ∼70% of cases.5,6 TP53 is characterized by at least 12 isoforms that share part of the DNA-binding domain and contain different transactivating and C-terminal domains.7 The majority of TP53 mutations consist of missense variants occurring in the protein DNA binding domain.5,8 Although most deletions are accompanied by a mutation in the remaining allele leading to a complete loss of wild-type (WT) TP53, heterozygous TP53 mutations are also common, as indicated by a median variant allele frequency (VAF) of typically 40% to 60%.4,9 In the context of CK AML, the presence of TP53 alterations further reduces patient overall survival to <5% at 3 years.4,5,8-10 Targeting recurrent molecular alterations, such as FLT3 mutations, with midostaurin represents an emerging strategy to improve patient outcome in AML11 ; however, this approach appears less conceivable for CK AML, because this subgroup is characterized by high genetic complexity and infrequently harbors targetable mutations.4
As part of the Leucegene project, our group developed a drug discovery platform for AML treatment. We first clinically and genetically (RNA sequencing) annotated a collection of 415 primary human AML specimens and developed an integrated bioinformatic platform for data analysis.12 Because cell lines do not recapitulate the complexity of human cancers, we then optimized ex vivo cell culture conditions that maintain leukemia stem cell activity,13 allowing us to perform high-throughput chemical screening on primary AML specimens. Integration of responses of these well-characterized specimens to a large collection of chemical compounds14-16 allowed us to uncover compound sensitivity profiles for several AML subtypes. In this paper, we report the successful application of this strategy to CK AML and reveal the central role of cell proliferation genes in this disease, along with the unique sensitivity of CK AML to polo-like kinase 1 (PLK1) inhibitors.
Methods
Study approval
The Leucegene project is an initiative approved by the Research Ethics Boards of Université de Montréal and Maisonneuve-Rosemont Hospital. All leukemia samples and paired normal DNA specimens were collected and characterized by the Quebec Leukemia Cell Bank after obtaining an institutional Research Ethics Board–approved protocol with informed consent according to the Declaration of Helsinki. The Quebec Leukemia Cell Bank is a biobank certified by the Canadian Tissue Repository Network.
Next-generation sequencing and mutation validations
Sequencing was performed as previously described.14 Sequence data were mapped to the reference genome hg19 according to RefSeq annotations (University of California, Santa Cruz, 16 April 2014). Variants were identified using CASAVA 1.8.2 or km (https://bitbucket.org/iric-soft/km) approaches. All variants present in genes mutated in myeloid cancers or in acute leukemias were investigated (supplemental Table 1). Acquired or germline origin of these variants not present in the COSMIC database was confirmed by Sanger sequencing of nontumoral DNA from mouth swabs or saliva. Other genes with recurrent variants (ie, in 5 or more complex AML samples) were also analyzed in nontumoral DNA. Genes and positions were also investigated by km approach as previously described, using a 5% VAF cutoff for missense and nonsense mutations as well as for indels confirmed by another approach, of 10% for other indels.17 Suspected TP53 mutations not meeting coverage criteria were all assessed by Sanger sequencing. In samples with low TP53 expression (<1 reads per kilobase million), TP53 exons 4 to 8 based on NM_00546 were also sequenced.
Cell culture and chemical screen
Cell cultures from frozen AML mononucleated cells and chemical screens were handled as previously described14 using serum-free media supplemented with cytokines, 500 nM SR1 (Alichem), and 500 nM UM729 (Institute for Research in Immunology and Cancer [IRIC]). Compounds were added to seeded cells in serial dilutions (8 dilutions, 1:3, 10 μM down to 4.5 nM for primary screen and 10 dilutions, 1:3, 3.33 μM down to 0.17 nM for validation screen) in duplicate wells. The exception was daunorubicin, for which dilutions from 1 μM to 0.45 nM were performed. Control wells received dimethyl sulfoxide (0.1%) only. Cell viability was evaluated after 6-day culture using the CellTiterGlo assay (Promega) according to the manufacturer’s instructions. Percentage of inhibition for dose response curves was calculated as 100 − (100 × (mean luminescence [compound]/mean luminescence [dimethyl sulfoxide])). Values of 50% inhibitory concentration (IC50) were calculated using ActivityBase SARview Suite. For cases where compounds failed to inhibit AML cell survival/proliferation, IC50 values were arbitrarily reported at the highest dose tested (10 μM).
Additional materials and methods can be found in the supplemental Material.
Results
The global Leucegene cohort is composed of 68 CK and 347 other “non-CK” AML specimens. The baseline characteristics of these 2 groups are provided in Figure 1A. As anticipated, CK AML was associated with older age, lower white blood cell counts, and inferior patient survival (median survival 3 vs 12 months, P < .001; Figure 1B).
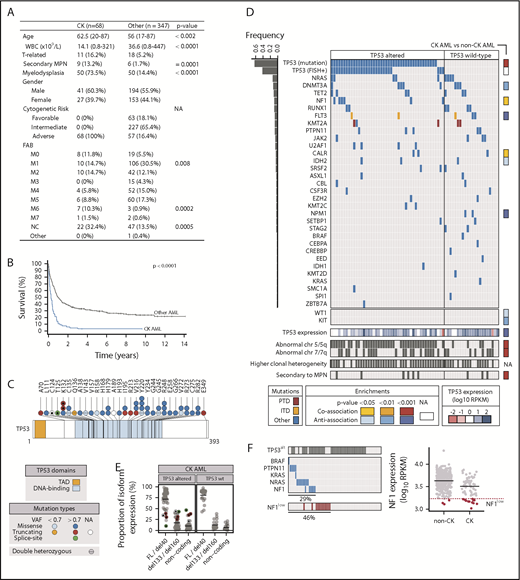
Clinical and genetic characteristics of CK AML cohort. (A) Clinical characteristics of CK and non-CK AML cohorts. (B) Patient survival comparing CK AML (n = 68) vs non-CK AML (n = 347). All patients with available survival data are represented, including those not treated with intensive chemotherapy. (C) Primary structure of TP53 and positions of mutations. (D) Mutations identified in CK AML. Each column represents a patient sample. CK AML samples are grouped according to their TP53 alteration status: altered (mutated and/or abnormal FISH) or not altered (no mutation and no deletion by FISH). Bar graph on the left indicates mutation frequency, and column on the right provides the enrichment for a mutation in CK vs non-CK cohorts. (E) TP53 isoform expression in CK AML. (F) Analysis of mutations in genes involved in the RAS pathway. CK samples with NF1 expression below the first percentile of control AML, designated by the dotted line, were considered to have low NF1 expression. P values are based on 2-tailed Fisher's exact test or on Wilcoxon rank-sum test. FAB, French-American-British; FL, full-length; ITD, internal tandem duplication; MPN, myeloproliferative neoplasm; NA, not applicable; NC, not classified; PTD, partial tandem duplication; RPKM, reads per kilobase million; TAD, transactivating domain; T-AML, therapy-related acute myeloid leukemia; WBC, white blood cell counts.
Clinical and genetic characteristics of CK AML cohort. (A) Clinical characteristics of CK and non-CK AML cohorts. (B) Patient survival comparing CK AML (n = 68) vs non-CK AML (n = 347). All patients with available survival data are represented, including those not treated with intensive chemotherapy. (C) Primary structure of TP53 and positions of mutations. (D) Mutations identified in CK AML. Each column represents a patient sample. CK AML samples are grouped according to their TP53 alteration status: altered (mutated and/or abnormal FISH) or not altered (no mutation and no deletion by FISH). Bar graph on the left indicates mutation frequency, and column on the right provides the enrichment for a mutation in CK vs non-CK cohorts. (E) TP53 isoform expression in CK AML. (F) Analysis of mutations in genes involved in the RAS pathway. CK samples with NF1 expression below the first percentile of control AML, designated by the dotted line, were considered to have low NF1 expression. P values are based on 2-tailed Fisher's exact test or on Wilcoxon rank-sum test. FAB, French-American-British; FL, full-length; ITD, internal tandem duplication; MPN, myeloproliferative neoplasm; NA, not applicable; NC, not classified; PTD, partial tandem duplication; RPKM, reads per kilobase million; TAD, transactivating domain; T-AML, therapy-related acute myeloid leukemia; WBC, white blood cell counts.
Characterization of TP53 allelic expression and transcript isoforms in CK AML
TP53 alterations (TP53alt), comprising either mutations (Figure 1C) or deletions (Figure 1D), as detected by RNA sequencing and fluorescent in situ hybridization (FISH), respectively, were found in 68% (46/68) of CK AML specimens analyzed and were associated with a trend toward inferior survival (supplemental Table 2; Figure 1D; supplemental Figure 1A). As reported, most of the TP53 point mutations observed in CK specimens of the Leucegene cohort were missense mutations located in the DNA-binding domain (Figure 1C), followed by truncating mutations, missense mutations outside the DNA binding domain, and splice-site mutations (supplemental Figure 1B). Among TP53alt samples, 54% (25/46) were detectable by RNA sequencing and FISH, 39% (18/46) by RNA-seq alone, and 7% (3/46) by FISH alone. Surprisingly, and in contrast to results from DNA sequencing studies,4,9 messenger RNA–based VAF values of ≥80% were observed in the majority of TP53mut specimens that show normal FISH (13/18). DNA sequencing of 3 of these 13 specimens for which genomic DNA and RNA was available confirmed the marked discordance between the genomic DNA in which WT and mutated alleles were easily detected vs the cDNA in which expression of the mutated allele was predominant (supplemental Figure 1C). This suggests some level of allele-specific expression of the mutated TP53 alleles. Supporting, this possibility, we found that 3 of the 18 patient specimens had compound heterozygous TP53 mutations for which the sum of VAF was >85%. Altogether, these results indicate that WT TP53 allelic expression is lost in most “FISH-negative” CK TP53alt AML cases. Notably, levels of TP53 are comparable or modestly lower in CK AML compared with other AML (supplemental Figure 1D). Two cases, however, showed strikingly reduced TP53 expression levels despite at least 1 TP53 allele was present as evaluated by FISH, suggesting that loss of TP53 expression can occur in a small proportion of cases that appears normal by FISH and mutation analyses.
When assessing TP53 isoform expression (n = 28 transcript isoforms in GRCh38.84 annotation), we found that in most AML samples, long isoforms of TP53, emanating from its canonical promoter and encoding for full-length protein isoforms, as well as those with a short N-terminal truncation (40 amino acids),7 are predominantly expressed (full-length/del40; Figure 1E; supplemental Figure 1E). In addition, we detected great variability in expression levels of transcripts that are driven from an internal TP53 promoter and that code for protein isoforms with large (133 or 160 amino acids) N-terminal truncations (del133/del160; Figure 1E). These shorter TP53 protein isoforms were previously shown to dampen TP53 function18 and promote breast cancer cell survival.19 Interestingly, we found that in cases with mutations in TP53 exons 4 and 5, expression ratios of long vs short and noncoding TP53 isoforms were markedly skewed against long TP53 protein isoforms (see highlighted dots in Figure 1E, corresponding to dots marked with a star in Figure 1C). This was not only the case in patient 11H002, carrying a splice-donor mutation in exon 4 (highlighted green dot in Figure 1E), but also in 2 patients with frameshift mutations affecting lysine 132, distal to the exon 5 splice-acceptor, as well as in a patient with a frameshift mutation in exon 4. These cases represent novel examples where coding or noncoding mutations have unexpected impacts on alternative splicing or promoter usage of TP53.
NF1/RAS pathway is frequently activated in CK AML
Thirty-one additional genes were mutated in CK AML (Figure 1D; supplemental Table 3), among them NRAS (9/68, 13%), DNMT3A (8/68, 12%), TET2 (7/68, 10%), NF1 and RUNX1 (6/68 each, 9%), FLT3, MLL/KMT2A and PTPN11 (5/68 each, 7%), JAK2 and U2AF1 (4/68 each, 6%), CALR, IDH2, and SRSF2 (3/68 each, 4%), among others shown in Figure 1D (frequency <3/68). In addition to TP53, CK AML specimens were significantly associated with NF1 (P = .02) and CALR (P = .01) mutations, and antiassociated with FLT3 (P = 2 × 10−9), DNMT3A (P = .004), KIT (P = .008), WT1 (P = .02), and IDH2 mutations (P = .04) when compared with other AML specimens in the Leucegene cohort (supplemental Table 4). In line with our results, NF1 loss was recently shown to be associated with adverse risk AML, where it confers an inferior clinical outcome.20 Altogether, 10% of CK AML specimens contained mutations in JAK2 or CALR, occurring predominantly (6/7) in cases with a documented history of myeloproliferative neoplasms (Figure 1D). Using an unbiased approach and a minimal frequency of 10%, no other recurrent mutations were detected in this cohort.
Of interest, at least 1 component of the RAS pathway (NF1, NRAS, KRAS, PTPN11, BRAF) is mutated in 29% (20/69) of CK specimens (Figure 1F left panel). Six RAS pathway mutated cases had a VAF < 25%, suggesting that these mutations frequently represent late occurring events in CK AML (supplemental Tables 3). NF1 encodes neurofibromin 1, a negative regulator of the RAS signaling pathway, and loss of NF1 results in Ras activation.21 Interestingly, NF1 is located on chromosome band 17q11.2, a region that is recurrently deleted in AML.22 Most (4/6) mutations in this gene consisted of nonsense or frameshift variants leading to truncated proteins. We also determined that NF1 expression levels are lower in CK AML compared with non–CK AML (P = 5 × 10−6) and that 19% (13/68) of CK AML samples have NF1 expression levels inferior to that of the first percentile of non–CK AML (NF1low; Figure 1F right). Although RAS-pathway mutations are less frequent in CK AML than in some other genetic subtypes of AML,4 and of these mutations, only NF1 is specifically enriched in CK AML, our results indicate that 46% of CK AML samples in our cohort (31/68) have either RAS pathway mutations or decreased NF1 expression (Figure 1F left), thus representing the second most dysregulated pathway in this AML subgroup.
Screening for compounds that preferentially target CK AML
In order to identify potent drugs targeting CK AML, we tested a collection of 233 small molecules (supplemental Table 5) enriched in clinical grade compounds that could be repurposed for this leukemia subtype. A total of 27 primary CK AML samples and 11 intermediate-risk control AML samples (Figure 2A; supplemental Table 6) were tested using our previously described AML cell culture conditions.13
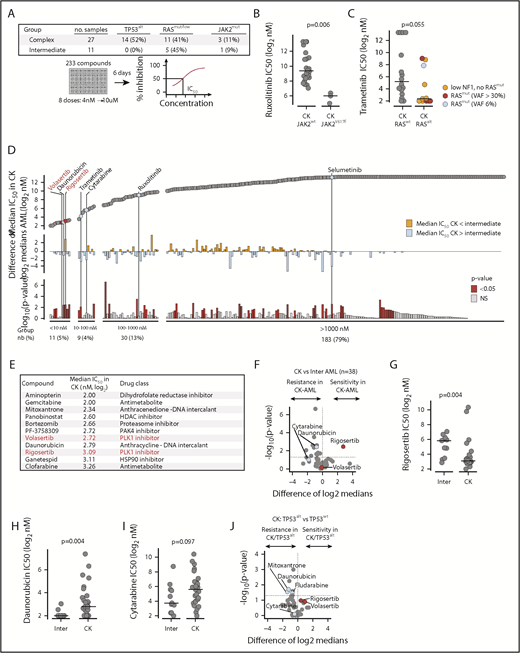
Drug repurposing screen to identify potent inhibitors of CK AML proliferation. (A) Layout of the discovery primary chemical screen. (B) Comparison of JAK1/2 inhibitor ruxolitinib activity in CK JAK2WT vs JAK2mut AML specimens. (C) Comparison of MEK1/2 inhibitor trametinib activity in CK RASWT vs RASalt AML. (D) Overview of the results of the discovery primary chemical screen. Compounds are ordered from the most active (left) to the least active (right) in CK AML. Upper panel shows median IC50 in CK AML; middle panel shows differential sensitivity in CK vs intermediate risk AML, and lower panel shows the statistical significance of these differences. PLK1 inhibitors are shown in red, and other selected compounds are shown in blue. (E) Most active compounds on CK AML are listed with their corresponding IC50 (Log2 nM) and targets. (F) Volcano plot showing the differential sensitivity in CK vs intermediate risk AML of compounds active in CK AML (IC50 < 1000 nM). Dot plot comparison of rigosertib (G), daunorubicin (H), and cytarabine (I) IC50 values (Log2 nM) between intermediate risk and CK AML. (J) Volcano plot showing the differential sensitivity in CK TP53WT vs TP53alt of compounds active in CK AML (IC50 < 1000 nM). In volcano plots, PLK1 inhibitors rigosertib and volasertib are depicted in red, whereas cytarabine and daunorubicin are depicted in blue. Medians are represented by a black line in dot plots. HDAC, histone deacetylase; nb, number; NS, not significant.
Drug repurposing screen to identify potent inhibitors of CK AML proliferation. (A) Layout of the discovery primary chemical screen. (B) Comparison of JAK1/2 inhibitor ruxolitinib activity in CK JAK2WT vs JAK2mut AML specimens. (C) Comparison of MEK1/2 inhibitor trametinib activity in CK RASWT vs RASalt AML. (D) Overview of the results of the discovery primary chemical screen. Compounds are ordered from the most active (left) to the least active (right) in CK AML. Upper panel shows median IC50 in CK AML; middle panel shows differential sensitivity in CK vs intermediate risk AML, and lower panel shows the statistical significance of these differences. PLK1 inhibitors are shown in red, and other selected compounds are shown in blue. (E) Most active compounds on CK AML are listed with their corresponding IC50 (Log2 nM) and targets. (F) Volcano plot showing the differential sensitivity in CK vs intermediate risk AML of compounds active in CK AML (IC50 < 1000 nM). Dot plot comparison of rigosertib (G), daunorubicin (H), and cytarabine (I) IC50 values (Log2 nM) between intermediate risk and CK AML. (J) Volcano plot showing the differential sensitivity in CK TP53WT vs TP53alt of compounds active in CK AML (IC50 < 1000 nM). In volcano plots, PLK1 inhibitors rigosertib and volasertib are depicted in red, whereas cytarabine and daunorubicin are depicted in blue. Medians are represented by a black line in dot plots. HDAC, histone deacetylase; nb, number; NS, not significant.
First, we correlated compound response to mutation status in CK AML and found a strong association between JAK2 mutations and sensitivity to compounds targeting JAK2 itself (ruxolitinib) or downstream JAK2 targets (mammalian target of rapamycin [rapamycin, temsirolimus]) as well as to a few selected kinase inhibitors (Figure 2B; supplemental Figure 2A-B). Samples with RAS pathway alterations showed an increased sensitivity to MEK inhibitors such as trametinib and selumetinib (Figure 2C; supplemental Figure 2A,C), similar to what we observed previously in MLL rearranged AML.14
In the second approach and irrespective of mutation status, we unbiasedly queried our compound collection to identify potent antiproliferative CK AML molecules. In this subgroup, 5% (11/233) of compounds were very potent (defined as median IC50 < 10 nM), whereas 4%, 13%, and 79% were active (10-100 nM), moderately active (100-1000 nM), or inactive (>1000 nM), respectively (Figure 2D; supplemental Table 7). Very active compounds (listed in Figure 2E) included several well-described agents already in use for the treatment of AML and other malignancies, such as the anthracyclines daunorubicin and mitoxantrone, the antimetabolites gemcitabine and clofarabine, and the dihydrofolate reductase inhibitor aminopterin. It also included the histone deacetylase inhibitor panobinostat, the proteasome inhibitor bortezomib, the HSP90 inhibitor ganetespib, the p21 protein activated kinase inhibitor PF-3758309, and, most strikingly, the 2 PLK1 inhibitors volasertib and rigosertib. Interestingly, HSP90 inhibitors have been shown to promote MDM2/CHIP E3 ligase-mediated degradation of mutant TP53 proteins23 (reviewed in Prokocimer et al24 ). HSP90 is also essential to PLK1 activity.25
CK AML specimens are particularly sensitive to PLK1 inhibition
The PLK1 inhibitor rigosertib was the only potent compound (ie, IC50 < 1000 nM in CK AML), which was more active in CK than in intermediate-risk AML (P = .0035) (Figure 2D,F-G). CK AML specimens were also very sensitive to volasertib, but sensitivity differences between subgroups could not be seen as volasertib’s IC50 reached the bottom limit of our dose response range (4 nM) (Figure 2F). In contrast to PLK1 inhibitors, the standard agents used for AML induction, namely cytarabine and daunorubicin, were less potent in CK AML, reflecting the lower efficacy of these agents in this AML subgroup (Figure 2F,H-J).
To confirm that PLK1 targeting is of particular interest in CK AML, we performed a validation screen on 29 primary AML samples (18 normal karyotype [NK] + 11 CK AML; supplemental Table 8) and used a lower dose-response range for volasertib (0.17 nM to 3.33 μM). Although volasertib is a highly selective PLK1 inhibitor, rigosertib is not, because it inhibits several other kinases, protein domains, and microtubule formation.26-28 Thus, we added the highly specific adenosine triphosphate (ATP)–competitive PLK1 inhibitor GSK461364 and the Aurora B inhibitor barasertib as an independent control (Figure 3A-B). Volasertib and GSK461364 showed strong antiproliferative activity against AML (median IC50: 7.9 nM and 4.9 nM, respectively; supplemental Table 9) with greater potency in the CK AML subgroup (Figure 3C). Interestingly, normal hematopoietic cells appear as sensitive as NK AML specimens, suggesting that a therapeutic window exists for CK AML targeting with these compounds. In agreement with their high specificity toward PLK1, response patterns of the 2 PLK1 inhibitors were highly correlated across AML samples tested, indicative of on-target activity for these 2 structurally unrelated compounds (Figure 3D, Pearson r correlation = 0.91). Barasertib29,30 had a low IC50 in AML (median IC50: 13.3 nM) but lacked specificity toward the CK group (Figure 3C) and showed a response pattern unrelated to that of the 2 PLK1 inhibitors (dotted line in Figure 3D; Pearson r correlation with volasertib = 0.21, Pearson r correlation with GSK461364 = 0.13). These results strongly suggest that PLK1 is an interesting therapeutic target in adverse cytogenetic AML, and that both volasertib and GSK461364 are worth investigating in this context.

Validation screen for PLK1 inhibitors. (A) PLK1 inhibitors used in primary and secondary screens along with Aurora B inhibitor barasertib are depicted with their respective structures and clinical advancement. (B) Layout of the validation screen assay. (C) Dot plot representation of volasertib, GSK461364, and barasertib IC50 values (Log2 nM) in normal hematopoietic cells (2 samples are CD34+ cord blood cells and 1 sample is CD34+ mobilized peripheral blood), NK, and CK AML samples. (D) Correlation analysis of inhibitory responses (Log2 IC50 in nM) across AML samples. (E) IC50 values (Log2 nM) of volasertib, GSK461364, and barasertib are compared in CK AML samples according to their TP53 status (WT vs altered). Medians are represented by a black line in dot plots. alt, altered; NK, normal karyotype; wt, wild type.
Validation screen for PLK1 inhibitors. (A) PLK1 inhibitors used in primary and secondary screens along with Aurora B inhibitor barasertib are depicted with their respective structures and clinical advancement. (B) Layout of the validation screen assay. (C) Dot plot representation of volasertib, GSK461364, and barasertib IC50 values (Log2 nM) in normal hematopoietic cells (2 samples are CD34+ cord blood cells and 1 sample is CD34+ mobilized peripheral blood), NK, and CK AML samples. (D) Correlation analysis of inhibitory responses (Log2 IC50 in nM) across AML samples. (E) IC50 values (Log2 nM) of volasertib, GSK461364, and barasertib are compared in CK AML samples according to their TP53 status (WT vs altered). Medians are represented by a black line in dot plots. alt, altered; NK, normal karyotype; wt, wild type.
TP53-altered CK AML samples are also highly responsive to PLK1 inhibitors
As expected, TP53alt CK AML samples were generally more resistant to compounds than TP53wt samples (Figure 2J). Moreover, TP53 alterations are responsible for a specific resistance to genotoxic agents and to drugs usually used in clinics (daunorubicin, fludarabine, mitoxantrone). In contrast, the 3 PLK1 inhibitors tested in our primary and secondary screens were as efficient on TP53alt specimens as on TP53wt CK AML (Figures 2J and 3E).
G2/M signature in CK AML and its correlation to PLK1 inhibition sensitivity
Interestingly, we observed that expression of marker of proliferation Ki-67 (MKI67) and PLK1 is higher in CK AML than in non-CK samples (Figure 4A-B), suggestive of a dysregulation in proliferation and/or cell-cycle progression. In line with this observation, PLK1 and MKI67 expression is significantly higher in bone marrow (BM) compared with peripheral blood cells, in accordance with the increased intrinsic proliferative activity of BM cells (supplemental Figure 3A-B). In both blood and BM AML samples, CK AML cells always showed higher PLK1 and MKI67 expression compared with non-CK (supplemental Figure 3B). Broader analysis of cell-cycle genes (GO:0007049) in human AML specimens of the full Leucegene cohort indicates that these genes are very strongly correlated to each other, representing the most correlated gene network (Figure 4C; supplemental Table 10). According to the predominant functions of PLK1 in G2/M transition and mitosis, we found that the G2/M checkpoint genes are significantly enriched in CK AML (Figure 4D; supplemental Tables 11 and 12). Clonal heterogeneity is a feature of a subgroup of CK AML and has been associated with inferior patient outcomes.31 Interestingly, higher clonal heterogeneity was associated with a stronger G2/M expression signature in CK AML samples (Figure 4E). In line with these observations, expression of G2/M checkpoint genes was used to reclassify samples included in the primary chemical screen (Figure 4F). Strikingly, high expression of the G2/M signature predicted an increased sensitivity to PLK1 inhibitors in CK AML samples (Figure 4G), revealing vulnerability in this adverse AML subtype.
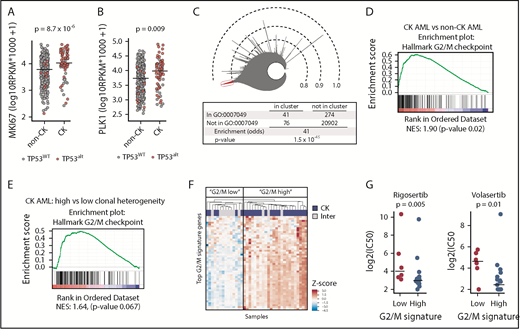
Higher expression of G2/M genes in CK AML and correlation to PLK1 inhibitors sensitivity.MKI67 (A) and PLK1 (B) messenger RNA expression in CK AML compared with non-CK. TP53WT samples are depicted in gray, and TP53alt samples are depicted in red. (C) Icicle representation of all correlated genes in the human AML transcriptome (cohort n = 415) showing that cell-cycle genes (GO:0007049) are highly enriched in the most correlated peak (red), thus indicating that cell-cycle genes are highly correlated in human AML. (D) Gene set enrichment analysis in CK (n = 68) compared with non-CK AML (n = 347) and showing results for the hallmark G2/M checkpoint gene set (supplemental Table 11), which was the single most enriched hallmark set. (E) Gene set enrichment analysis in CK AML comparing samples with higher (n = 55) vs low (n = 13) clonal heterogeneity. Results show the hallmark G2/M checkpoint gene set. (F) Hierarchical clustering performed on samples included in the primary screen using the 50 most enriched genes in hallmark G2/M checkpoint gene set. z scores were applied on rows. (G) Dot plot comparison of rigosertib and volasertib IC50 values (Log2 nM) restricted to CK AML samples classified according to their G2/M signature. Medians are represented by a black line. Inter, intermediate; NES, normalized enrichment score.
Higher expression of G2/M genes in CK AML and correlation to PLK1 inhibitors sensitivity.MKI67 (A) and PLK1 (B) messenger RNA expression in CK AML compared with non-CK. TP53WT samples are depicted in gray, and TP53alt samples are depicted in red. (C) Icicle representation of all correlated genes in the human AML transcriptome (cohort n = 415) showing that cell-cycle genes (GO:0007049) are highly enriched in the most correlated peak (red), thus indicating that cell-cycle genes are highly correlated in human AML. (D) Gene set enrichment analysis in CK (n = 68) compared with non-CK AML (n = 347) and showing results for the hallmark G2/M checkpoint gene set (supplemental Table 11), which was the single most enriched hallmark set. (E) Gene set enrichment analysis in CK AML comparing samples with higher (n = 55) vs low (n = 13) clonal heterogeneity. Results show the hallmark G2/M checkpoint gene set. (F) Hierarchical clustering performed on samples included in the primary screen using the 50 most enriched genes in hallmark G2/M checkpoint gene set. z scores were applied on rows. (G) Dot plot comparison of rigosertib and volasertib IC50 values (Log2 nM) restricted to CK AML samples classified according to their G2/M signature. Medians are represented by a black line. Inter, intermediate; NES, normalized enrichment score.
The PLK1 inhibitor volasertib delays the development of leukemia in vivo
We next assessed the antileukemic activity of volasertib in a xenotransplantation mouse model of human AML. First, NSG mice were transplanted with CK TP53-null HL-60 cells and treated with either volasertib or the conventional chemotherapeutic agent cytarabine (Figure 5A). At the tested doses, treatments were well tolerated as reflected by the absence of body weight loss in any of the conditions (supplemental Figure 4A). The HL-60 model is very aggressive, as control mice became ill within 5 to 6 weeks after transplantation, and cytarabine treatment failed to delay leukemia development in vivo (Figure 5B). Importantly, volasertib treatment significantly delayed the development of the disease reflected by an increase in survival of the treated mice (Figure 5B).
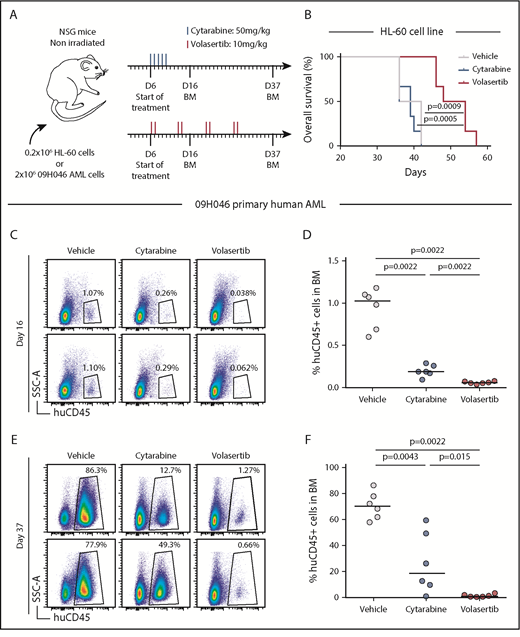
The PLK1 inhibitor volasertib delays development of poor outcome human AML in a xenotransplantation mouse model. (A) Summary of the treatment protocols and time point analysis for NSG mice transplanted with 0.2 million HL-60 leukemic cells or 2 million 09H046 AML cells. Six days after transplantation, mice were treated with either volasertib (IV, 2 times a week for 4 weeks at 10 mg/kg) or cytarabine (intraperitoneally for 5 consecutive days at 50 mg/kg). Vehicle-treated mice received both phosphate-buffered saline (intraperitoneally) and 0.9% NaCl (IV). BM aspirations were performed at days 16 and 37 in 09H046-transplanted mice only. (B) Kaplan-Meier curves as determined for vehicle-, cytarabine-, and volasertib-treated mice transplanted with HL-60 cells. Log-rank test was used to evaluate the statistical significance. Representative flow cytometry profiles of BM cells at day 16 (C) and day 37 (E) in vehicle-, cytarabine-, and volasertib-treated mice transplanted with 09H046 AML cells. (D,F) report the percentage of human CD45+ cells in BM at days 16 and 37 in all treated mice transplanted with 09H046 AML cells. Medians are represented by a black line in dot plots. SSC-A, side scatter area.
The PLK1 inhibitor volasertib delays development of poor outcome human AML in a xenotransplantation mouse model. (A) Summary of the treatment protocols and time point analysis for NSG mice transplanted with 0.2 million HL-60 leukemic cells or 2 million 09H046 AML cells. Six days after transplantation, mice were treated with either volasertib (IV, 2 times a week for 4 weeks at 10 mg/kg) or cytarabine (intraperitoneally for 5 consecutive days at 50 mg/kg). Vehicle-treated mice received both phosphate-buffered saline (intraperitoneally) and 0.9% NaCl (IV). BM aspirations were performed at days 16 and 37 in 09H046-transplanted mice only. (B) Kaplan-Meier curves as determined for vehicle-, cytarabine-, and volasertib-treated mice transplanted with HL-60 cells. Log-rank test was used to evaluate the statistical significance. Representative flow cytometry profiles of BM cells at day 16 (C) and day 37 (E) in vehicle-, cytarabine-, and volasertib-treated mice transplanted with 09H046 AML cells. (D,F) report the percentage of human CD45+ cells in BM at days 16 and 37 in all treated mice transplanted with 09H046 AML cells. Medians are represented by a black line in dot plots. SSC-A, side scatter area.
Second, NSG mice were transplanted with cells from the adverse outcome human AML sample 09H046. This primary specimen is characterized by chromosome 17 monosomy, TP53 mutation, a high frequency of leukemic stem cells,32 and displays a G2/M transcriptomic signature. As for the previous experiment, mice were treated with well-tolerated drug doses (Figure 5A; supplemental Figure 4B). This less aggressive model allowed us to monitor leukemia development in mice as assessed by determination of the percentage of human CD45+ cells in BM. At day 16 and 37 after transplantation, treated mice showed significant reduction of leukemic cells in BM compared with control mice (Figure 5C-F). Although the 09H046 model responded positively to cytarabine treatment, volasertib-treated mice achieved a more pronounced delay in leukemia development. Strikingly, median human CD45+ cells in BM at day 37 represented 70.2% in vehicle, 19.5% in cytarabine, and only 0.9% in volasertib-treated mice (median values; Figure 5E-F). Together, our data showed a strong anti-AML activity of volasertib in xenotransplantation mouse models of poor outcome human AML.
Discussion
We provide the analysis of the largest collection of CK AML specimens analyzed by RNA sequencing to date. This study confirms the very high occurrence of TP53 alterations in CK AML and supports previous findings indicating that screening for TP53 mutations must be performed to complement cytogenetic studies in clinical laboratories. Strikingly, TP53 VAF determined by RNA sequencing in our cohort was particularly high compared with previously reported next-generation targeted DNA sequencing-based studies,4,9 and further investigations revealed that WT TP53 allelic expression is underrepresented in most CK TP53alt AML cases in our cohort. This supports the hypothesis that loss of functional TP53 is an important contributor to AML disease progression, especially following therapy-induced selection.
The therapeutic strategy for high-risk AML patients is composed of chemotherapy combining daunorubicin and cytarabine, followed by allogeneic hematopoietic stem cell transplantation. However, this approach fails to induce durable remission in the vast majority of CK AML patients. This prompted us to conduct a drug repurposing screen using primary AML samples characterized by RNA sequencing. We found that targeting JAK2 mutations was the most promising mutation-based approach in the CK subgroup. In line with these findings, JAK2 inhibition has been shown to provide clinical benefits in some AML patients with previous myeloproliferative neoplasms.33 Most importantly, our screen identified the PLK1 inhibitors rigosertib, volasertib, and GSK461364 as potent drugs that preferentially target proliferation/survival of CK AML cells.
PLK1 is a serine/threonine kinase essential for G2/M transition as well as many steps of the mitotic process, including centrosome maturation, spindle assembly, inactivation of the anaphase-promoting complex, and mitotic exit.34 PLK1 phosphorylates hundreds of proteins, including the antiapoptotic protein Bcl-x(L) and mTORC1, suggesting that PLK1 functions extend beyond mitosis.35,36 PLK1 activity is increased in tissues and cells with a high mitotic index, and its expression is upregulated in various cancers.37-42 Interestingly, our transcriptomic data revealed an increased expression of several G2/M cell-cycle genes, including PLK1 and of the proliferative marker MKI67 in CK AML, suggestive of a dysregulation in proliferation and/or cell-cycle progression. Accordingly, BM samples express higher levels of cell-cycle genes and MKI67 due to increased proliferative activity of these cells compared with peripheral blood specimens43 ; however, CK samples expressed higher levels of PLK1 irrespective of the sample source. Strikingly, high G2/M signature predicts an increased sensitivity to PLK1 inhibitors, highlighting vulnerability in CK AML cells. On the other hand, inhibition of Aurora B, which is predominantly active during mitosis and thus operates after the G2/M checkpoint, did not show any specificity toward CK AML. This further suggests that G2/M transition, precisely, is a therapeutic target for CK AML.
Several PLK1 inhibitors have been developed, and some are under clinical evaluation for solid tumors and AML treatment (Figure 3A; reviewed in Liu and Talati et al 44,45 ). The PLK1 inhibitor volasertib acquired Food and Drug Administration breakthrough therapy status in 2013 for AML. A recent phase 2 clinical trial was conducted in older AML patients ineligible for remission induction therapy, evaluating their response to low-dose cytarabine with or without volasertib.46 Volasertib combination therapy increased the overall response rate in their cohort composed of various risk groups. An active randomized phase 3 trial (NCT01721876) is currently prospectively assessing the value of this regimen similarly in older AML patients. Preliminary results revealed that a major obstacle to the addition of volasertib at the tested dose in this frail population is the increased frequency of lethal infections,47 highlighting the need to balance toxicities in the light of expected benefits. Our in vitro experiments suggest that a therapeutic window exists for CK AML targeting, which appeared more sensitive to PLK1 inhibition than NK AML specimens and normal hematopoietic cells. In addition, in vivo xenotransplantation mouse models of poor outcome human AML also confirmed the strong anti-AML activity of volasertib.
Volasertib48 and GSK46136449,50 are selective ATP-competitive inhibitors of PLK1, whereas rigosertib is a non-ATP competitive inhibitor proposed to also target the RAS pathway and multiple kinases.26-28,51 However, results of our primary AML screen showed no specificity of rigosertib related to the RAS pathway status. Anti-AML activity of rigosertib correlated rather with that of volasertib, suggesting that PLK1 is the target at play in AML. Similarly, high correlation in response rate between the structurally different volasertib and GSK461364 compounds also suggests that they act through PLK1 inhibition in AML. In a wide variety of cells, PLK1 inhibition results in cell proliferation defects, cell-cycle arrest at the G2/M phase, and spindle abnormalities followed by apoptotic cell death.52,53 Autophagy induced by PLK1 inhibition is another mechanism reported for anti-AML activity.54 Meanwhile, activity of PLK1 inhibitors on bromodomain proteins has been documented, but its contribution to antileukemic activity warrants further investigation.55
Because of the crosstalk between PLK1 and the p53 tumor suppressor in cell-cycle regulation and DNA damage pathways, some studies have suggested that TP53alt cancer cells would be more sensitive to PLK1 inhibition.56,57 In our screens, TP53alt primary AML were as sensitive to PLK1 inhibitors as their TP53wt counterparts, indicating that PLK1 targeting is efficient at altering proliferation/survival of these refractory cells in vitro.
CK and TP53alt AML have a very unfavorable outcome even with intensive therapies. Our results, demonstrating the preferential activity of PLK1 inhibitors in this subgroup, provide a rationale to rapidly test these agents in CK AML clinical trials.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Muriel Draoui for project coordination, Vincent Archambault and Jana Krosl for insightful discussions, Marie-Ève Bordeleau and Tara Macrae for correcting the manuscript, Marianne Arteau and Raphaëlle Lambert at the IRIC genomics platform for RNA sequencing as well as Jean Duchaine, Sébastien Guiral, Karine Audette, and Caroline Gauvin at the IRIC high-throughput screening platform for chemical screens. The collaboration of BCLQ coinvestigators and the dedicated work of BCLQ staff, namely Giovanni d’Angelo and Claude Rondeau, are also acknowledged.
This work was supported by the Government of Canada through Genome Canada and the Ministère de l’Économie, de l’Innovation et des Exportations du Québec through Génome Québec, with supplementary funds from AmorChem. Support from the Leukemia Lymphoma Society and Canadian Cancer Society Research Institute (G.S.) is also acknowledged. J.H. holds a research chair from Industrielle-Alliance (Université de Montréal). BCLQ is supported by grants from the Cancer Research Network of the Fonds de Recherche du Québec–Santé. RNA-Seq read mapping and transcript quantification were performed on the supercomputer Briaree from Université de Montréal, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation, NanoQuébec, RMGA, and the Fonds de Recherche du Québec–Nature et Technologies (FRQ-NT). C.T. and V.-P.L. are supported by a fellowship from the Cole Foundation, and V.-P.L. is a Vanier Canada Graduate Scholarship fellow. C.M. is a recipient of a CIHR fellowship (MFE-148251).
Authorship
Contribution: C.M. contributed to project conception, performed and analyzed chemical screens and in vivo experiments, generated corresponding material, and cowrote the manuscript; V.-P.L. contributed to the project conception, analyzed genomic data and chemical screens, generated corresponding material, and cowrote the manuscript; C.T. contributed to project conception and performed and analyzed chemical screens; B.L. performed TP53 isoform analysis; I.B. contributed to chemical screen and data validation; N.M., M.F., V.B.-C., and S.C. helped carry out in vivo experiments; Y.G. formulated volasertib for in vivo administration; S. Lavallée performed FISH experiments and prepared the karyotypes; S. Lemieux was responsible for supervision of the bioinformatics team and the statistical analyses; A.M. contributed to selection of compounds and is responsible for the chemistry team as part of the Leucegene project; J.H. contributed to project conception, analyzed the cytogenetic and FISH studies, selected AML samples, and provided clinical data; and G.S. contributed to project conception and coordination and cowrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Guy Sauvageau, Institute for Research in Immunology and Cancer, P.O. Box 6128, Station Centre-Ville, Montreal, QC H3C 3J7, Canada; e-mail: guy.sauvageau@umontreal.ca; and Josée Hébert, Banque de Cellules Leucémiques du Québec, 5415 L'Assomption Blvd, Montreal, QC H1T 2M4, Canada; e-mail: josee.hebert@umontreal.ca.

