

Key Points
Individuals harboring biallelic HAVCR2 germline mutations are highly susceptible to sporadic primary SPTCL.
Somatic mutations in the genes involved in epigenetic regulation/signal transduction might be associated with clonal expansion of SPTCL.

Abstract
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare subtype of peripheral T-cell lymphoma affecting younger patients and associated with hemophagocytic lymphohistiocytosis. To clarify the molecular pathogenesis of SPTCL, we analyzed paired tumor and germline DNAs from 13 patients by whole-exome sequencing. All cases were Asians and were phenotypically sporadic with no family history of SPTCL. Consistent with a recent report, germline mutations in HAVCR2, encoding T-cell immunoglobulin mucin 3 (TIM3), were identified in 11 of 13 (85%) cases. All mutated cases were primary SPTCL, whereas the 2 cases without mutation were secondary SPTCL associated with underlying diseases, including viral infection and autoimmune disease. Ten patients harbored homozygous p.Y82C mutations, and 1 showed compound heterozygous mutations (p.Y82C and p.T101I). Both missense mutations altered highly conserved residues located in the extracellular immunoglobulin variable–like domain. According to the Genome Aggregation Database of >138 500 general individuals, both mutations were documented with minor allele frequencies < 0.007, indicating remarkable enrichment of these HAVCR2 alleles in SPTCL. SPTCL cells also harbored somatic mutations (6.2 per patient) that are frequently identified in genes associated with epigenetic regulation and signal transduction. In conclusion, individuals harboring biallelic HAVCR2 (TIM3) germline mutations were highly susceptible to sporadic SPTCL, which was also associated with clonal somatic mutations.
Introduction
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is an uncommon subtype of peripheral T-cell lymphoma predominantly affecting younger individuals.1-3 SPTCL patients present with multifocal subcutaneous nodules and sometimes with life-threatening hemophagocytic lymphohistiocytosis (HLH). Immunosuppressive therapy yields good outcomes in SPTCL, unlike other lymphomas, which require chemotherapy. SPTCL is relatively common in women, but no racial or ethnic predisposition has been reported.2 Although somatic mutations have been reported in genes associated with epigenetic modifiers and the PI3K/AKT/mTOR pathway,4 none of the genes were frequently mutated in SPTCL cases. Therefore, the molecular pathogenesis of SPTCL is still poorly understood. To elucidate the causative genetic background and clonal lesions in SPTCL, we explored the spectrum of germline and somatic mutations in SPTCL cases by whole-exome sequencing (WES).
Methods
Patient enrollment
In this study, 13 patients in whom SPTCL was diagnosed by hematopathologists reviewing biopsy samples of subcutaneous nodules with immunohistochemistry, were enrolled from Thailand and Japan (Table 1). All samples were collected with informed consent following approval of the institutional review boards of the respective institutions. Using the previously published protocol, clonality of tumor cells was assessed by polymerase chain reaction analysis of T-cell receptor (TCR) gene rearrangement.5
Clinical, ethnic, and genetic characteristics of enrolled patients with SPTCL
| UPN | Sex | Age, y | Ethnicity | Family history of SPTCL | Skin lesion | Fever | HLH | Underlying disease | Treatment | Outcome | TCR rearrangement | HAVCR2 germline mutation | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aa change | Allelic status | ||||||||||||
| 1 | M | 37 | Asian | No | Buccal | No | No | N.P. | CHOP/CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 2 | F | 30 | Asian | No | Upper limbs | No | No | N.P. | PSL/CsA | Relapse | Monoclonal | Y82C/T101I | Compound heterozygous |
| 3 | F | 41 | Asian | No | Generalized | Yes | No | N.P. | CsA | CR | Oligoclonal | Y82C | Homozygous |
| 4 | F | 35 | Asian | No* | Trunk/eyelid | No | No | N.P. | CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 5 | F | 26 | Asian | No | Generalized | Yes | Yes | N.P. | EPOCH | Relapse | Monoclonal | Y82C | Homozygous |
| 6 | F | 32 | Asian | No | Trunk | No | No | N.P. | CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 7 | M | 24 | Asian | No | Trunk | Yes | No | N.P. | CsA | CR | Monoclonal | Y82C | Homozygous |
| 8 | M | 28 | Asian | No | Trunk | No | No | N.P. | CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 9 | F | 27 | Asian | No | Lower limb | No | Yes | HIV infection | CsA | Dead | N.A. | Wild-type | N.A. |
| 10 | F | 5 | Asian | No | Lower limbs | Yes | No | N.P. | PSL/CsA | CR | Monoclonal | Y82C | Homozygous |
| 11 | F | 46 | Asian | No | Generalized | Yes | Yes | SLE | CsA | Relapse | Monoclonal | Wild-type | N.A. |
| 12 | F | 59 | Asian | No | Eyelid | Yes | Yes | N.P. | CsA | PR | Monoclonal | Y82C | Homozygous |
| 13 | M | 33 | Asian | No | Trunk | No | No | N.P. | CsA | Relapse | N.A. | Y82C | Homozygous |
| UPN | Sex | Age, y | Ethnicity | Family history of SPTCL | Skin lesion | Fever | HLH | Underlying disease | Treatment | Outcome | TCR rearrangement | HAVCR2 germline mutation | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aa change | Allelic status | ||||||||||||
| 1 | M | 37 | Asian | No | Buccal | No | No | N.P. | CHOP/CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 2 | F | 30 | Asian | No | Upper limbs | No | No | N.P. | PSL/CsA | Relapse | Monoclonal | Y82C/T101I | Compound heterozygous |
| 3 | F | 41 | Asian | No | Generalized | Yes | No | N.P. | CsA | CR | Oligoclonal | Y82C | Homozygous |
| 4 | F | 35 | Asian | No* | Trunk/eyelid | No | No | N.P. | CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 5 | F | 26 | Asian | No | Generalized | Yes | Yes | N.P. | EPOCH | Relapse | Monoclonal | Y82C | Homozygous |
| 6 | F | 32 | Asian | No | Trunk | No | No | N.P. | CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 7 | M | 24 | Asian | No | Trunk | Yes | No | N.P. | CsA | CR | Monoclonal | Y82C | Homozygous |
| 8 | M | 28 | Asian | No | Trunk | No | No | N.P. | CsA | Relapse | Monoclonal | Y82C | Homozygous |
| 9 | F | 27 | Asian | No | Lower limb | No | Yes | HIV infection | CsA | Dead | N.A. | Wild-type | N.A. |
| 10 | F | 5 | Asian | No | Lower limbs | Yes | No | N.P. | PSL/CsA | CR | Monoclonal | Y82C | Homozygous |
| 11 | F | 46 | Asian | No | Generalized | Yes | Yes | SLE | CsA | Relapse | Monoclonal | Wild-type | N.A. |
| 12 | F | 59 | Asian | No | Eyelid | Yes | Yes | N.P. | CsA | PR | Monoclonal | Y82C | Homozygous |
| 13 | M | 33 | Asian | No | Trunk | No | No | N.P. | CsA | Relapse | N.A. | Y82C | Homozygous |
aa, amino acid; CHOP, cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulfate (Oncovin), and prednisone; CR, complete remission; CsA, cyclosporin A; EPOCH, etoposide phosphate, prednisone, vincristine sulfate (Oncovin), cyclophosphamide, doxorubicin hydrochloride (hydroxydaunomycin); F, female; M, male; N.A., not assessed; N.P., nothing particular; PR, partial remission; PSL, prednisolone; UPN, unique patient number.
A cousin developed nodal lymphoma of unknown type.
Extraction of DNA
DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tissue (n = 12), fresh frozen tissue (n = 1), or fresh bone marrow/peripheral blood (n = 13) using a DNeasy Tissue Kit, a Gentra Puregene Blood Kit, or a GeneRead DNA FFPE Kit (all from QIAGEN), according to the manufacturer’s protocols.
WES and confirmatory deep sequencing
For WES, paired tumor (fresh frozen and FFPE tissues) and germline (bone marrow or peripheral blood) samples were collected from all patients. WES libraries were prepared from 100 to 500 ng of genomic DNA and generated using a SureSelect Human All Exon V6 kit (Agilent Technologies), followed by massively parallel sequencing of enriched exon fragments on a HiSeq 2500 with 125–base pair paired-end mode, as previously described.6,7 Following the manufacturers’ protocols, libraries from FFPE samples were prepared using a KAPA Hyper Prep Kit (Kapa Biosystems) or a SureSelect Low Input Automated Target Enrichment System (Agilent Technologies). Mutations were called using the in-house pipeline Genomon2 (version 2.5.2.) and EBCall, as previously described.8,9 Variants located at the simple repeat site registered in the UCSC database (http://hgdownload.cse.ucsc.edu/goldenPath/hg38/database/) were excluded from further analysis. Integrative Genomics Viewer (IGV) software10 version 2.4.14 was used for the manual inspection of sequencing results. For validation, amplicon-based deep sequencing was applied to candidate variants, as previously described.6,11 To further assess accumulation of germline variants in SPTCL, global allele frequencies in representative variants were extracted from the Genome Aggregation Database (gnomAD) (downloaded on 16 October 2018).12 The WES data were deposited in the European Genome-phenome Archive database (accession number EGAS00001003282).
Computational analysis
Statistical analyses were performed using R (http://www.R-project.org). Multiple significance testing was adjusted according to the method described by Hochberg and Benjamini.13 The methods used for the statistical analyses are described in detail in “Results.” HAVCR2 mutations were assessed by in silico analyses using PolyPhen-214 and Combined Annotation Dependent Depletion.15
Results
Accelerated immunity and inflammation with cytotoxic CD8+/TCRαβ+ T-cell involvement in SPTCL
The median age of diagnosis in the 13 patients was 32 years (range, 5-59) (Table 1). All enrolled patients were phenotypically sporadic without family history of SPTCL or malignant lymphoma of the skin. Among them, 11 had primary SPTCL without any underlying disease, whereas the other 2 were assumed to have secondary SPTCL related to HIV infection and systemic lupus erythematosus (SLE) (Table 1). Multiple SPTCL lesions were commonly observed on the trunks, eyelids, extremities (Figure 1A), and buccal mucosa. Pathologically, atypical lymphocytes infiltrated into subcutaneous adipose tissue with karyorrhexis, and adipocytes were rimmed by these lymphocytes (Figure 1B). These atypical lymphocytes were positive for CD3, CD5, CD8, TIA1, and βF1 (TCRβ subunit) by immunohistochemical staining, indicating a cytotoxic phenotype of tumor cells and their potential interaction with antigen-presenting cells. All SPTCL patients responded to immunosuppressive therapy; HLH was present in 4 patients (30.8%), suggesting that SPTCL is deeply associated with accelerated immunity and inflammation.

Individuals with biallelic HAVCR2 germline mutations are highly susceptible to SPTCL. (A) Representative images of a case with SPTCL. Multiple subcutaneous nodules with erythema diffusely involved lower legs (upper left panel). A positron emission tomography/computed tomography scan showed multiple regions with 18 fluorodeoxyglucose (FDG) uptake in subcutaneous soft tissue (yellow arrows). (B) Representative pathological images of SPTCL. On hematoxylin and eosin staining, infiltrating lymphocytes with cellular atypia-rimmed adipocytes (upper left panel). On immunohistochemical staining, these atypical cells were positive for CD3, CD8, and βF1 (TCRβF1) (lower left and right panels). Scale bars, 50 µm. (C) Germline mutations in HAVCR2 (NM_032782), encoding TIM3 protein, detected by WES. Representative images from IGV software showed homozygous p.Y82C mutation in UPN3 (left panel) and compound heterozygous p.Y82C and p.T101I mutations in UPN2 (right panel). In these images, mutated nucleotides (c.245A>G and c.302C>T as shown by reverse complementary DNA sequence) are highlighted in blue and green on sequence reads (horizontal gray bars), respectively. Larger black boxes outline the codons affected by mutations, and the smaller black boxes contain the number of bases with/without substitution, as indicated by the corresponding colors. Functional domains of TIM3, together with sites and zygosities of HAVCR2 mutations, are also shown (middle panel). Among 13 patients, 10 harbored homozygous p.Y82C (blue circles), and 1 patient harbored heterozygous p.Y82C and p.T101I (red circles). (D) Homology of the mutated residues in TIM3 protein. Alignment of amino acid sequences in immunoglobulin variable–like domain of TIM3 among different species is shown (Homo sapiens, Gorilla gorilla gorilla, Mus musculus, Rattus norvegicus, Bos taurus, Pan troglodytes, Canis lupus, and Macaca mulatta). Two residues of TIM3 mutated in SPTCL were highly conserved, as highlighted in red (Y82) and green (T101). (E) Enrichment of HAVCR2 germline mutations (p.Y82C) in SPTCL. Allele frequencies (left panel) and homozygote ratios (right panel) of p.Y82C (c.245A>G) are shown in SPTCL patients from our study, as well as from general populations from the gnomAD dataset. SPTCL patients, East Asians, South Asians, and the whole general population are indicated in red, blue, green, and purple, respectively. *P < .001, Fisher’s exact test.
Individuals with biallelic HAVCR2 germline mutations are highly susceptible to SPTCL. (A) Representative images of a case with SPTCL. Multiple subcutaneous nodules with erythema diffusely involved lower legs (upper left panel). A positron emission tomography/computed tomography scan showed multiple regions with 18 fluorodeoxyglucose (FDG) uptake in subcutaneous soft tissue (yellow arrows). (B) Representative pathological images of SPTCL. On hematoxylin and eosin staining, infiltrating lymphocytes with cellular atypia-rimmed adipocytes (upper left panel). On immunohistochemical staining, these atypical cells were positive for CD3, CD8, and βF1 (TCRβF1) (lower left and right panels). Scale bars, 50 µm. (C) Germline mutations in HAVCR2 (NM_032782), encoding TIM3 protein, detected by WES. Representative images from IGV software showed homozygous p.Y82C mutation in UPN3 (left panel) and compound heterozygous p.Y82C and p.T101I mutations in UPN2 (right panel). In these images, mutated nucleotides (c.245A>G and c.302C>T as shown by reverse complementary DNA sequence) are highlighted in blue and green on sequence reads (horizontal gray bars), respectively. Larger black boxes outline the codons affected by mutations, and the smaller black boxes contain the number of bases with/without substitution, as indicated by the corresponding colors. Functional domains of TIM3, together with sites and zygosities of HAVCR2 mutations, are also shown (middle panel). Among 13 patients, 10 harbored homozygous p.Y82C (blue circles), and 1 patient harbored heterozygous p.Y82C and p.T101I (red circles). (D) Homology of the mutated residues in TIM3 protein. Alignment of amino acid sequences in immunoglobulin variable–like domain of TIM3 among different species is shown (Homo sapiens, Gorilla gorilla gorilla, Mus musculus, Rattus norvegicus, Bos taurus, Pan troglodytes, Canis lupus, and Macaca mulatta). Two residues of TIM3 mutated in SPTCL were highly conserved, as highlighted in red (Y82) and green (T101). (E) Enrichment of HAVCR2 germline mutations (p.Y82C) in SPTCL. Allele frequencies (left panel) and homozygote ratios (right panel) of p.Y82C (c.245A>G) are shown in SPTCL patients from our study, as well as from general populations from the gnomAD dataset. SPTCL patients, East Asians, South Asians, and the whole general population are indicated in red, blue, green, and purple, respectively. *P < .001, Fisher’s exact test.
Biallelic germline mutations in HAVCR2, encoding TIM3, are positive for primary sporadic SPTCL
In WES, the median percentages of target regions per patient covered at depths of ≥30, ≥20, ≥10, and ≥2 read counts were 87.6%, 91.1%, 93.9%, and 96.5%, respectively, and the median sequence depth was 101.8. Compatible with the phenotype of SPTCL, as mentioned above, among the 13 patients, 11 patients (84.6%) showed germline missense mutations in HAVCR2 (NM_032782) (chr5:156 512 843-156 536 248; hg19), encoding T-cell immunoglobulin mucin 3 (TIM3) in tumor and corresponding germline samples (Figure 1C; Table 1). Substitutions in the mutations were c.245A>G (p.Y82C; NP_116171) or c.302C>T (p.T101I), which affected highly conserved residues among species (Figure 1D). In silico analysis showed that p.Y82C and p.T101I mutations were predicted to be deleterious at scores of 1 and 0.993, respectively, in PolyPhen-2, and at scores of 20.2 and 15.08, respectively, in Combined Annotation Dependent Depletion.
Clinically, all 11 patients with HAVCR2 mutations were primary SPTCL cases, whereas the 2 cases without mutations were secondary SPTCL associated with HIV infection and SLE (Table 1). Accordingly, 100% of primary SPTCL cases in this study were positive for germline mutations. Among them, 10 cases were positive for the homozygous p.Y82C mutation, which was identical to that reported in a recent publication of familial SPTCL cases.16 The other case was positive for p.Y82C and p.T101I, which were located on the independent alleles, resulting in a compound heterozygous configuration confirmed by IGV software (Figure 1C). As for another underlying disease, HAVCR2 mutations were identified in the SPTCL patients with HLH, as well as in those without HLH, which was in line with the results published.16
Individuals with germline HAVCR2 mutations are highly susceptible to SPTCL
Although the p.Y82C mutation is a rare germline allele in the Asian population,16 p.T101I is a novel variant in a patient of southeast Asian (Thai) origin (UPN2). Among the general population in the gnomAD dataset, the global minor allele frequencies (MAFs) of p.Y82C (rs184868814) and p.T101I (rs147827860) were 3.6 × 10−3 (n = 138 558) and 6.6 × 10−3 (n = 138 607), respectively (Figure 1E). Although p.Y82C is more prevalent in East Asians (n = 9434; MAF = 2.1 × 10−2), p.T101I is more frequent in South Asians (n = 15 391; MAF = 1.8 × 10−3) than in East Asians (MAF = 0). Therefore, a p.T101I allele in UPN2 is likely to be inherited from a South Asian ancestor. The frequency of p.Y82C homozygotes was 76.9% in our SPTCL cohort (10 of 13 patients) and 0.0029% in the general population (4 of 1.4 × 105), resulting in the remarkable enrichment of the p.Y82C homozygote in SPTCL (odds ratio [OR], 1.2 × 105; 95% confidence interval [CI], 2.2 × 104 to 5.8 × 105). With regard to MAF, the OR for a p.Y82C allele is 1.2 × 103 (95% CI, 4.4 × 102 to 3.1 × 102) in SPTCL. Further analysis of each ethnicity, including East Asians and South Asians, showed significant enrichment in East Asians (OR, 1.0 × 104; 95% CI, 1.8 × 103 to 5.8 × 104 [for homozygote] and OR, 2.0 × 102; 95% CI, 7.3 × 10 to 5.2 × 102 [for MAF]) and in combined East and South Asian cohorts (OR, 2.8 × 104; 95% CI, 5.0 × 103 to 1.5 × 105 [for homozygote] and OR, 4.6 × 102; 95% CI, 1.7 × 102 to 1.2 × 102 [for MAF]), suggesting clear susceptibility of subjects with germline HAVCR2 mutations to SPTCL.
Recurrent somatic mutations in epigenetic regulation and signal transduction are positive in SPTCL
By WES analysis of SPTCL and matched germline samples, we identified 80 somatic mutations (range, 0-48; mean, 6.2; median, 2.0) in 13 patients (supplemental Table 1). We selected 3 mutations (in AP2A1, DHX29, and FADD) for validation that were associated with somatic mutations reported in a previous SPTCL study (Figure 2).4 The sequencing depth was ≥500 read counts (range, 511-18 366 per patient; median, 4062 per patient), and all 3 mutations were successfully validated by amplicon-based deep sequencing. The median value of the variant allelic frequencies was 0.14, suggesting the obvious clonal expansion of SPTCL cells. By combining our results with those from the previous study,4 58 genes were recurrently mutated in SPTCL patients and are primarily involved in epigenetic modification or the PI3K/AKT/mTOR pathway, including TET2, CHD3, CHD4, ARID1B, and others (Figures 2 and 3). These results suggest that additional somatic mutations might be associated with SPTCL genesis, as well as germline predisposition.
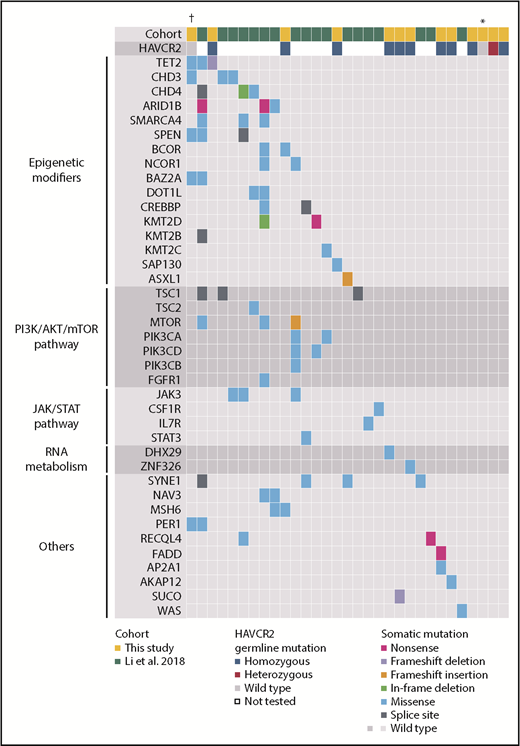
Landscape of somatic and germline mutations in SPTCL. On the basis of combined results from a previous study4 and this study (supplemental Table 1), the spectrum of somatic and germline mutations in SPTCL is shown. Each column represents an individual case, and each row represents the indicated mutated gene. Genes were assigned to the functional categories shown on the left. The types and status of the mutations are shown by the indicated colors. †Case with HIV infection. *Case with SLE.
Landscape of somatic and germline mutations in SPTCL. On the basis of combined results from a previous study4 and this study (supplemental Table 1), the spectrum of somatic and germline mutations in SPTCL is shown. Each column represents an individual case, and each row represents the indicated mutated gene. Genes were assigned to the functional categories shown on the left. The types and status of the mutations are shown by the indicated colors. †Case with HIV infection. *Case with SLE.
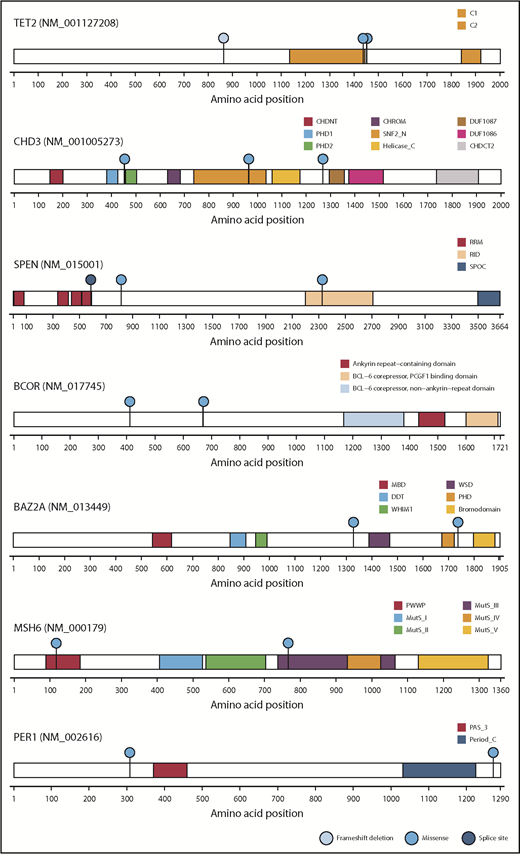
Genes mutated in common between the independent cohorts of SPTCL. Among the entire spectrum of mutations (Figure 2; supplemental Table 1), mutations in TET2, CHD3, BCOR, BAZ2A, SPEN, MSH6, and PER1 are shown, because mutations in these genes were identified in a previous study4 and in this study. Functional domains of the mutated genes and the types of mutations are indicated by the corresponding colors.
Genes mutated in common between the independent cohorts of SPTCL. Among the entire spectrum of mutations (Figure 2; supplemental Table 1), mutations in TET2, CHD3, BCOR, BAZ2A, SPEN, MSH6, and PER1 are shown, because mutations in these genes were identified in a previous study4 and in this study. Functional domains of the mutated genes and the types of mutations are indicated by the corresponding colors.
Discussion
TIM3 is a transmembrane protein expressed by CD8+ T cells and natural killer cells,17-19 and it acts as a negative immune checkpoint through interactions with cognate ligands, such as galectin-9.4,20 The TIM3 with p.Y82C mutation was shown to induce protein misfolding and abrogate expression of this molecule on the plasma membrane, leading to the loss of its function promoting acceleration of immunity and inflammation in SPTCL with HLH.16 This may explain the pathophysiology of SPTCL: unlike other lymphomas, immunosuppressive therapy for SPTCL yields good outcomes, and HLH is frequently observed. By contrast, TIM3 is activated in stem cells of acute myeloid leukemia and regulates their self-renewal potential.21,22 Therefore, tumorigenesis due to loss of TIM3 function is likely to be relevant in a tissue-specific manner.
HLH is a major complication in SPTCL. Although TIM3-mutant SPTCL patients were reported to have HLH in a previous study (14/16; 87.5%),16 this complication was less frequent in TIM3 mutants in our cohort (2/11; 18.2%). According to previous reports, the risk of developing HLH in SPTCL patients is 6% to 20%,2,23 which is more typical of our study than that by Gayden et al.16 The discrepancy in the frequencies of HLH in these cohorts may be associated with variation in the patients’ backgrounds. Before sequencing, the background prevalence of HLH in the entire cohort (with and without HAVCR2 mutation) was already lower (4/13; 30.8%) in our study than in the study by Gayden et al16 (14/27; 51.9%). The other reason that could have led to the discrepancy is the median age of patients with an HAVCR2 mutation: 32 years vs 26 years, respectively. The speculation that defects in TIM3 result in HLH is plausible; however, SPTCL in itself is also significantly associated with HAVCR2 mutations, irrespective of HLH, which is a common conclusion in both studies. So far, a study with a large number of SPTCL patients of various age groups and HAVCR2 mutational status has not been reported. To clarify the incidence of HLH in HAVCR2-mutated patients, further analysis of a larger cohort is warranted.
According to the previous studies, SPTCL is associated with autoimmune and infectious diseases.24,25 For example, SPTCL cases with HIV infection were reported previously,26,27 although T-cell lymphoma is reported to be relatively rare in HIV-infected patients.28 In fact, a patient in this study harbored HIV infection as an underlying disease. In this patient, multiple somatic mutations in genes associated with epigenetic modification and signal transduction, but no germline mutation in HAVCR2, were detected. The other patient without an HAVCR2 germline mutation showed accelerated autoimmunity due to SLE. These findings suggest that somatic mutations and underlying diseases are associated with SPTCL pathogenesis, in addition to germline risk.
SPTCL is pathologically diagnosed on the basis of neoplastic infiltration of CD8+ and TCR α/β+ T cells into subcutaneous tissue. Now that Gayden et al16 and we showed that SPTCL is a genetically distinct disease that is characterized by highly frequent HAVCR2 mutation, genotyping of HAVCR2 will provide additional information for diagnosis of this disease. Future presentation of SPTCL might be predicted in family members of primary patients. As for the usefulness of HAVCR2 genotyping for differential diagnosis of other T-cell lymphomas and familial HLH syndromes, further study is required.
In conclusion, SPTCL is a congenital disease that is caused by biallelic germline HAVCR2 mutations, and it shows an autosomal recessive–inherited pattern. In addition to these germline mutations, recurrent somatic mutations in the genes involved in epigenetic regulation and signal transduction might be associated with the pathogenesis of SPTCL.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Center for Anatomical, Pathological and Forensic Medical Research, Kyoto University Graduate School of Medicine, for preparing microscope slides, Jerasit Surintrspanont for taking pathological images, and Ravi Velaga for English editing. The authors also thank gnomAD and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about.
This work was supported by the Thailand Research Fund (RSA6080011) (C.P.); Research Collaborations in Hematologic Malignancies and Hematopoietic Stem Cell Transplantation, Chulalongkorn University (U.B.); the Thailand Research Fund (IRG5780017) (C.P.), Grants-in-Aid from the Ministry of Health, Labor and Welfare of Japan and KAKENHI 23249052, 22134006, 21790907, and 15H05909 (S.O.) and 16H05338 (H.M.); project for the development of innovative research on cancer therapies (p-direct) (S.O.); the Japan Society for the Promotion of Science through the “Funding Program for World-Leading Innovative R&D on Science and Technology,” initiated by the Council for Science and Technology Policy (S.O.); Japan Society for the Promotion of Science Core-to-Core Program, A. Advanced Research Networks (H.M. and S.O.); project for cancer research and therapeutics evolution JP16cm0106501 (S.O.); the Project for Development of Innovative Research on Cancer Therapeutics 15cm0106056h0005 and 16cm0106501h0001 (S.O.); JP17km0305018 from the Japan Agency for Medical Research and Development (H.M.); the High Performance Computing Infrastructure System Research Project (hp150232) (S.O.); and research grants from the Friends of Leukemia Research Fund, the Princess Takamatsu Cancer Research Fund, the Takeda Science Foundation, The Japanese Society of Hematology, The Public Trust Fund for Clinical Cancer Research, and the SENSHIN Medical Research Foundation (H.M.).
Authorship
Contribution: C.P. collected samples and clinical data, extracted DNA, performed WES, analyzed data, and wrote the manuscript; Y.T. extracted DNA, performed WES, analyzed data, and wrote the manuscript; N.K., K.Y., H.S., Y.N., Y.S., K.C., H.T., and S.M. analyzed WES data; T.A. reviewed pathology reports; W.S. prepared and extracted DNA; U.B., A.P., K.W., P.L., N.U., S.K., C.M., M.S., and N.A. provided patient samples and clinical data; J.T. extracted DNA; Y.F. performed WES; P.R., S.O., and H.M. conceptualized the overall research and wrote the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hideki Makishima, Pathology and Tumor Biology, University of Kyoto, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8510, Japan; e-mail: makishima.hideki.8x@kyoto-u.ac.jp; Seishi Ogawa, Pathology and Tumor Biology, University of Kyoto, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8510, Japan; e-mail: sogawa-tky@umin.ac.jp; and Ponlapat Rojnuckarin, Department of Medicine, Faculty of Medicine, Chulalongkorn University, King Chulalongkorn Memorial Hospital, Bangkok 10330, Thailand; e-mail: rojnuckarinp@gmail.com and ponlapat.r@chula.ac.th.

