

Key Points
ATL cells evade the senescence response triggered by HTLV-1 Tax-mediated NF-κB hyperactivation.
ATL cells have acquired senescence-mitigating genetic/epigenetic changes.

Abstract
Human T-cell leukemia virus type 1 (HTLV-1) is the etiological agent of adult T-cell leukemia/lymphoma (ATL). The HTLV-1 viral trans-activator/oncoprotein Tax is a major driver of ATL, yet it induces rapid p21Cip1/Waf1 (p21)- and p27Kip1-mediated cellular senescence through constitutive activation (hyperactivation) of NF-κB. Although constitutive NF-κB activation is a common feature of T/B-cell leukemia/lymphoma, including ATL, it is not known how ATL cells maintain chronic NF-κB activation without undergoing senescence. Here, we demonstrate that, in contrast to HTLV-1− T-cell lines, ATL cell lines no longer undergo Tax-induced senescence. Although Tax+ and Tax− ATL cell lines showed signatures of constitutive NF-κB activation, their ability to progress through the cell cycle was unaffected. In some cases, ATL cell lines continued to proliferate despite significant upregulation of p21; additionally, many cell lines displayed altered expression of G1 and G1/S cyclins, particularly overexpression of cyclin D2. We propose that, during the course of ATL development, leukemia cells acquire genetic/epigenetic changes that can mitigate the senescence response triggered by NF-κB hyperactivation. Restoring the NF-κB–induced senescence response would likely help to control the development and progression of ATL and similar lymphoid malignancies.
Introduction
Human T-cell leukemia virus type 1 (HTLV-1) is the etiological agent of adult T-cell leukemia/lymphoma (ATL). The HTLV-1 trans-activator/oncoprotein Tax is a potent activator of IKK/NF-κB,1 the principal driver of HTLV-1 leukemogenesis.2,3 Tax also promotes genomic instability and cell-cycle abnormalities.4,5 Although Tax is an oncogene, its expression counterintuitively induces a rapid senescence response mediated by tumor suppressors p21Cip1/Waf1 (p21) and p27Kip1 (p27).6 We have previously shown that the senescence response induced by Tax is caused by potent and persistent NF-κB activation (termed “NF-κB hyperactivation”),7 a common feature of ATL and other leukemia/lymphomas.8-12 This Tax-driven dysregulation of the NF-κB pathway causes increased expression of NF-κB–regulated genes. If NF-κB hyperactivation, be it Tax driven or not, triggers a senescence response, then leukemia/lymphoma cells addicted to chronic NF-κB activation must have acquired genetic/epigenetic changes that can prevent or attenuate senescence induction to allow them to reach their leukemogenic potential. Here, we examine multiple T-cell lines of ATL origin for supporting evidence. Our results indicate that, although Tax expression in HTLV-1− T-cell lines leads to cell-cycle arrest/senescence, persistent Tax expression in Tax+ ATL cells or re-expression of Tax in Tax− ATL cells fails to trigger a similar senescence response. Additionally, significant p21 upregulation, cyclin D2 overexpression, and p27 downregulation are frequently observed in ATL cells that evade cell cycle arrest/senescence. The possible causes for the senescence-resistant phenotype of ATL are discussed.
Materials and methods
Cell culture
Human T-cell lines were cultured in RPMI 1640 (HyClone) supplemented with 10% fetal bovine serum (FBS), l-glutamine, 100 U/mL penicillin, and streptomycin and maintained in 5% CO2 at 37°C. ATL-55T cells were grown in the presence of 10 U/mL human interleukin-2 (Sigma-Aldrich). HeLa/18x21-EGFP cells, used as immunoblot control, were grown in Dulbecco’s modified Eagle medium (Quality Biologicals) with 10% FBS.
DNA transfection
The piggyBac transposon plasmids piggyBac–18x21-EGFP reporter and piggyBac-Tax were described previously.13 Lipofection and calcium phosphate–mediated DNA transfection were used to transfect T cells and HEK-293T cells, respectively.
Lentiviral vectors
Lentiviral vectors for Tax and the 18x21-EGFP reporter were derived as previously described.7,14 They were generated by cotransfecting HEK-293T cells with the vectors, along with 2 packaging plasmids that direct the expression of HIV structural proteins or the G glycoprotein of the vesicular stomatitis virus. The lentiviruses were added to the media, and cells were spinoculated at 1200g for 2 hours at 25°C; cells were then allowed to grow undisturbed for 24 hours. Tax and the reporter were introduced into ATL-55T (IL+) cells using the piggyBac transposon system; the 2 constructs were cloned into piggyBac vectors and transfected using FuGENE HD (Promega).
Growing tax-transduced T cells in semisolid medium
Methyl cellulose solution (4% weight-to-volume ratio; Sigma M-0512) was autoclaved and cooled to 4°C. The solution was then mixed with an equal volume of 2× RPMI 1640 medium (HyClone; 2× RPMI 1640 + 20% FBS + penicillin/streptomycin + glutathione) to make a 2% semisolid stock medium, which was kept in solution at 4°C. Plates were overlaid with 1% semisolid medium, and pelleted T cells transduced with lentiviral vectors for Tax and the 18x21-EGFP reporter were resuspended in a small volume of liquid RPMI 1640 and added dropwise to the overlaid plates. The embedded cells were grown at 37°C in a CO2 incubator, where the semisolid medium solidified. Green fluorescent protein (GFP)+ T cells were monitored for 7 to 10 days.
Immunoblotting
Cells were harvested and lysed in lysis buffer (Cell Signaling). Standard methods were used for immunoblotting. Samples containing 25 μg of whole-cell proteins, as determined by BCA assay (Promega), were loaded on 13.5% Tris-glycine gels. The HTLV-1 Tax mouse hybridoma monoclonal antibody 4C5 was generated in our laboratory, as previously described.6 All other antibodies were commercially available and were obtained from Cell Signaling Technology [cyclin D1 (DCS6, #2926), cyclin D2 (D52F9) #3741, cyclin D3 (DCS22) #2936, cyclin E (HE12) #4129, Cdc2 (POH1) #9116, p16 ink4A #4824, p21 waf1/Cip1 (DCS60) #2946, p27 kip1 (SX53G8.5) #3698, phosphorylated (p)-IκBα S32/36 (5A5) #9246, and IκBα (L35A5) #4814], Santa Cruz Biotechnology [β-actin (C4) sc-47778, CDK4 (DCS-35) sc-23896, CDK6 (C-21) sc-177, RelB sc-48379, NF-κB p52 (c-5) sc-7386], and Advanced Bioscience Laboratories (HTLV-1 p24 #4310).
Results
ATL cells evade Tax-induced cell-cycle arrest/senescence response
To test the hypothesis that, during leukemia/lymphoma development, ATL cells have acquired genetic/epigenetic changes that render them resistant to the senescence response induced by HTLV-1 Tax-driven or Tax-independent NF-κB hyperactivation, we examined multiple T-cell lines of ATL origin for evidence of impairment to the Tax/NF-κB–induced senescence response. Eleven human T-cell lines were examined for their response to Tax. ATL-43, ATL-55T (interleukin-2 dependent), ED, MT-1, and TL-Om1 are Tax− ATL cell lines. ATL-2 and ATL-T are Tax+, as detected by Tax-reporter assay and p24-Gag expression (Figure 1, bottom left and middle panels; Figure 2A, lanes 9-10). MT-4 is an HTLV-1–transformed and Tax+ T-cell line (Figure 2A, lane 11) derived from infection in cell culture. Three HTLV-1− CD4+ T lymphoblastic leukemia cell lines (CEM, Jurkat, and Sup-T1) were included for comparison. We transduced the cell lines with a tax lentivirus vector along with a Tax-responsive reporter (18x21-EGFP).14 Integration of both genes leads to Tax expression and reporter activation, causing cells to express large amounts of GFP.14 Cell lines that endogenously express Tax were transduced with only the reporter vector. ATL-55T cells could not be efficiently transduced by lentivirus vectors; as such, PiggyBac transposon vectors were used to integrate tax and 18x21-EGFP genes.15 Neither method succeeded in the ATL-43 cell line; for that reason, it was excluded from further analysis. After dual Tax and 18x21-EGFP transduction, Tax-expressing GFP+ cells were monitored for 7 to 10 days. Cells were grown in semisolid media to ensure that the clusters of GFP+ cells were derived from the proliferation of single Tax+ cells and not from an aggregate of independent GFP+ cells. Whole-cell lysates of the T-cell lines were also examined for signatures of activation of the canonical and noncanonical NF-κB pathways, including p-IκBα, RelB, and p52 (a processing product of p100), as well as for markers of cell-cycle progression.
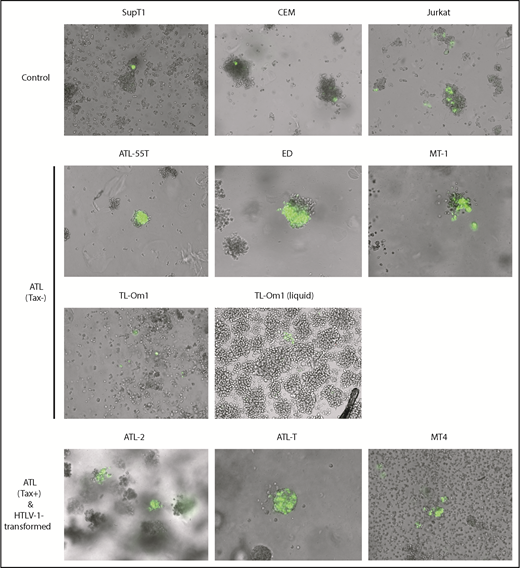
ATL cells are resistant to Tax-induced senescence. T cells were transduced with the HTLV-1 oncogenic protein Tax and an EGFP Tax-reporter plasmid14 and allowed to grow undisturbed for 7 to 10 days. Transduced T cells were monitored for proliferation in semisolid media, as described in “Materials and methods.” This experiment was repeated 3 times; representative images acquired using a 10× objective are shown.
ATL cells are resistant to Tax-induced senescence. T cells were transduced with the HTLV-1 oncogenic protein Tax and an EGFP Tax-reporter plasmid14 and allowed to grow undisturbed for 7 to 10 days. Transduced T cells were monitored for proliferation in semisolid media, as described in “Materials and methods.” This experiment was repeated 3 times; representative images acquired using a 10× objective are shown.
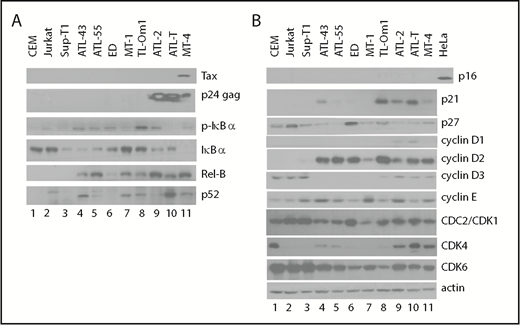
NF-κB activation and cell-cycle dysregulation in ATL and control T cells. Whole cell lysates were prepared as reported6 and analyzed by standard immunoblotting using the indicated antibodies. (A) Evaluation of NF-κB pathway activation. (B) Evaluation of cyclin-dependent kinase inhibitor, cyclin, and CDK expression. Each immunoblot shown used the same protein lysates; the β-actin control in panel B is applicable to panel A. Each blot was repeated ≥5 times with the same and different lysates.
NF-κB activation and cell-cycle dysregulation in ATL and control T cells. Whole cell lysates were prepared as reported6 and analyzed by standard immunoblotting using the indicated antibodies. (A) Evaluation of NF-κB pathway activation. (B) Evaluation of cyclin-dependent kinase inhibitor, cyclin, and CDK expression. Each immunoblot shown used the same protein lysates; the β-actin control in panel B is applicable to panel A. Each blot was repeated ≥5 times with the same and different lysates.
As shown in Figure 1, only single GFP+ cells could be seen in Sup-T1 and CEM controls (top left and middle panels) due to Tax-induced cell-cycle arrest/senescence, as previously reported.16 Small clusters of GFP+ cells were seen alongside individual GFP+ cells in Jurkat control cells (Figure 1, top right panel); however, the cell clusters were small as a result of limited cell division post-tax transduction. In contrast, large clusters of GFP+ cells were observed in ATL-55T, ED, and MT-1 cell lines after transduction of tax and 18x21-EGFP, indicating evasion of Tax-induced senescence (Figure 1, second row). This was also observed in TL-Om1 cells in liquid media but was less apparent in semisolid media (Figure 1, third row, right and left panels, respectively). As expected, Tax+ ATL-2, ATL-T, and MT-4 cell lines expressed abundant GFP after reporter transduction and continued to proliferate (Figure 1, bottom row). These results indicate that Tax+ and Tax− ATL cell lines, along with HTLV-1–transformed T-cell lines, no longer undergo senescence in response to Tax-driven NF-κB hyperactivation.
Constitutive NF-κB activation and cell-cycle dysregulation in ATL cell lines
After HTLV-1 infection progresses to ATL, leukemic cells in most cases (>60%) cease to express Tax.17 This is likely due to host cytotoxic T lymphocyte killing of Tax+ cells.18 Lack of Tax expression may allow ATL cells to evade immune surveillance, enabling clonal proliferation and expansion.19 Tax-triggered cellular senescence may also favor cells with low/no Tax expression.20 Importantly, ATL cells often constitutively express the HTLV-1 anti-sense mRNA-encoded bZIP protein, HBZ,21-25 which antagonizes many functions of Tax and Rex5,20 and promotes cell survival and proliferation.26,27 In the absence of Tax expression, ATL cells evolve chronic Tax-independent NF-κB hyperactivation.25 As such, we compared the state of NF-κB signaling in ATL cell lines with that in HTLV-1− T cells. As indicated by the immunoblot in Figure 2A, in contrast to the HTLV-1− CEM, Jurkat, and Sup-T1 cell lines, all ATL cell lines expressed p-IκBα (ATL-43, ATL-55T, ED, TL-Om1, ATL-2, MT4; lanes 4, 5, 6, 8, 9, and 11, respectively) or p52 (ATL-43, MT-1, TL-Om1, ATL-T, and MT-4; lanes 4, 7, 8, 10, and 11, respectively), signatures of activation of the canonical and noncanonical NF-κB pathways, respectively. In MT-4 cells, with the exception of a low level of p-IκBα, much of IκBα was degraded (Figure 2A, lane 11, compare rows 3 and 4). The expression of RelB, which is induced by NF-κB RelA/p50, c-Rel, and Tax,7 was highly elevated in Tax+ ATL-2, ATL-T, and MT-4 cell lines (Figure 2A, lanes 9-11) and increased in all but 1 of the ATL cell lines (ED; Figure 2A, compare lane 6 with lanes 4-5 and lanes 7-10), further indicating NF-κB activation. That either or both of the NF-κB pathways are chronically activated in the ATL cell lines correlates with their resistance to Tax/NF-κB hyperactivation-driven senescence and their ability to exploit NF-κB for proliferation and survival.
Chronic NF-κB activation has been linked to loss of G1/S cell-cycle checkpoints and overexpression of the cyclin D (D1, D2, D3) and E families of protein. The senescence response to NF-κB hyperactivation is mediated by 2 cyclin-dependent kinase inhibitors (CDKIs), p21 and p27, in a p53- and pRb-independent manner.6,7 For cellular transformation and ATL development to occur after initial HTLV-1 infection, senescence checkpoints must be inactivated so that proliferation may continue despite Tax expression. To investigate the molecular basis of the cellular changes in ATL cells responsible for the loss of the senescence response, we assessed the ATL whole-cell lysates for expression levels of G1/S cyclins, G1 cyclin-dependent kinases (CDKs), and CDKIs.
As a whole, neither ATL cells nor HTLV-1− CEM, Jurkat, and Sup-T1 control cell lines show expression of CDKI p16 (Figure 2B, top row). In control T cells, the absence of NF-κB activation correlates with a lack of p21 expression (lanes 1-3). p21 expression, which is potently induced by Tax/NF-κB,6,28,29 is elevated in ATL43, TL-Om1, ATL-2, ATL-T, and MT-4 cells (Figure 2B, lanes 4 and 8-11, respectively) but is low or undetectable in ATL-55, ED, and MT-1 cells (lanes 5-7, respectively). Most ATL cell lines (ATL43, ATL55, MT-1, ATL-2, and ATL-T; Figure 2B, lanes 4, 5, 7, 9, and 10, respectively) and the HTLV-1–transformed MT-4 cell line (lane 11) have reduced levels of p27 compared with controls (lanes 1-3). Cyclin D1 is not typically expressed in T cells and could only be detected at low levels in ATL-2 and ATL-T cells (Figure 2B, lanes 9-10, respectively). Interestingly, cyclin D2 is significantly upregulated in all ATL and HTLV-1–transformed cell lines (Figure 2B, lanes 4-11) but is undetectable in HTLV-1− controls (lanes 1-3). In contrast, cyclin D3 is easily detected in the control T-cell lines (Figure 2B, lanes 1-3) but is expressed at low or undetectable levels in all ATL cell lines and MT-4 cells (Figure 2B, lanes 4-11). CDK1 and CDK6 are expressed in all T-cell lines, whereas CDK4 is differentially expressed in a subset (high: CEM, ATL-2, ALT-T, MT-4, Figure 2B [lanes 1, 9-11, respectively]; and low: ATL43 and ATL-55, Figure 2B [lanes 4-5, respectively]). Finally, no consistent alteration to cyclin E expression was seen in ATL cell lines. In aggregate, chronic NF-κB activation, cyclin D2 overexpression, frequent p27 downregulation, and significant p21 upregulation in the absence of cell-cycle arrest/senescence are common in ATL cells.
Expression levels of p21 and p27 varied among the ATL cells examined, suggesting that not all ATL cells evade senescence through the same mechanism. Interestingly, the TL-Om1 cell line expresses higher combined levels of p21 and p27 than the other ATL cells (Figure 2B, lane 8, rows 2 and 3) and is the least resistant to Tax-induced senescence; it is able to proliferate in the presence of Tax in liquid but not semisolid media. The reason for this phenotype is still unknown.
Discussion
The human retrovirus HTLV-1 causes ATL in vivo and transforms primary human T lymphocytes in cell culture. Because primary ATL cells are difficult to propagate in culture, our study is based on ATL cell lines. The HTLV-1 oncoprotein Tax potently activates viral transcription along with cell signaling pathways, such as NF-κB. It has been proposed that the dysregulation of NF-κB activation stimulates infected T cells to proliferate, resulting in ATL.2,25,30 Counterintuitively, we have found that Tax expression or HTLV-1 infection in lymphoid and nonlymphoid cells leads rapidly (within 1 cell cycle) to cell-cycle arrest/senescence (termed “Tax-induced rapid senescence” [Tax-IRS]) in an NF-κB–dependent manner.7,31 Here, we show that, although HTLV-1− T-cell lines undergo cell-cycle arrest/senescence upon Tax expression, ATL cell lines are largely impervious to Tax-IRS upon re-expression or persistent expression of Tax. Examination of markers of NF-κB activation and cell-cycle regulation in ATL cells reveals constitutive activation of the canonical or noncanonical NF-κB pathway, or both. In these cells, significant dysregulation of cell-cycle regulators, including cyclin D2 overexpression, p27 downregulation, and p21 upregulation, was observed in the absence of cell-cycle arrest/senescence.
We have previously shown that ectopic expression of the HTLV-1 HBZ protein allows HeLa cells to express Tax stably, albeit at a low level.7 Furthermore, although HTLV-1 infection in HeLa cells largely led to senescence, a small fraction (1%-2%) of infected cells continued to express Tax at low levels. In these cells, the activities of Tax and Rex were blocked by HBZ.7,20 Because ATL cells express HBZ constitutively, the senescence-resistant phenotype of ATL cells may be attributed in part to HBZ. Experiments are in progress to test this hypothesis.
A recent study has shown that HBZ transactivates the basic leucine zipper ATF-like transcription factor 3, which forms a transcriptional complex with another transcription factor, interferon response factor 4 (IRF4), to promote ATL cell gene expression and proliferation.27 Remarkably, the ablation of HBZ via CRISPR-Cas9 reduced the viability of ATL cells.27 It is unclear whether the loss of cell viability is due to the senescence response associated with NF-κB hyperactivation. Importantly, IRF4 gene amplification and gain-of-function mutations frequently occur in ATL.25,32,33 One gain-of-function mutation, K59R, has been shown to augment the transcriptional activity of IRF4.33 Whether the genetic alterations in IRF4 collaborate with HBZ to promote senescence evasion, T-cell transformation, and ATL oncogenesis remains to be determined.
Although HBZ can antagonize Tax-IRS in HTLV-1–infected cells that express low levels of Tax, the majority of infected cells become senescent, regardless of HBZ expression.20 As such, we think it is possible that T lymphocytes that no longer undergo Tax-induced senescence due to genetic or epigenetic alterations clonally expand after HTLV-1 infection. We further speculate that this loss of a senescence response facilitates the clonal expansion of infected cells, as well as the development of Tax-independent NF-κB hyperactivation in ATL cells (see visual abstract). We believe that some of the genetic alterations that mitigate NF-κB–induced senescence may be found in the recurrent gain-of-function mutations (eg, IRF4 mutations) identified by recent whole-genome analyses of ATL25 and that restoration of the senescence response may help to control the development and progression of ATL. Future studies will explore these possibilities.
Acknowledgments
The authors thank Masao Matsuoka for providing the ATL cell lines used in this study.
This work was supported by National Institutes of Health, National Cancer Institute grants R01CA140963 and R21CA216660.
Authorship
Contribution: A.M.D.S. and C.-Z.G. conceived and designed the experiments, wrote the manuscript, and approved the final submission; and A.M.D.S. performed the experiments and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Chou-Zen Giam, Department of Microbiology and Immunology, Uniformed Services University of the Health Sciences, 4301 Jones Bridge Rd, Bethesda, MD 20814; e-mail: chou-zen.giam@usuhs.edu.

