

Key Points
ELMO1 regulates GPVI-mediated platelet signaling through binding to RhoG.
ELMO1 negatively regulates platelet spreading.

Abstract
Phosphatidylinositol 3-kinase is an important signaling molecule that, once activated, leads to the generation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3). We performed a proteomic screen to identify PIP3-interacting proteins in human platelets. Among these proteins, we found engulfment and cell motility 1 (ELMO1), a scaffold protein with no catalytic activity. ELMO1 is expressed in platelets and interacts with active RhoG. However, the function of ELMO1 in platelets is not known. The focus of this study was to determine the function of ELMO1 in platelets utilizing ELMO1−/− mice. Platelet aggregation, granule secretion, integrin αIIbβ3 activation, and thromboxane generation were enhanced in ELMO1−/− platelets in response to glycoprotein VI (GPVI) agonists but unaltered when a protease-activated receptor 4 agonist was used. The kinetics of spreading on immobilized fibrinogen was enhanced in ELMO1−/− platelets compared with wild-type (WT) littermate controls. This suggests that ELMO1 plays a role downstream of the GPVI and integrin αIIbβ3 pathway. Furthermore, whole blood from ELMO1−/− mice perfused over collagen exhibited enhanced thrombus formation compared with WT littermate controls. ELMO1−/− mice showed reduced survival compared with control following pulmonary embolism. ELMO1−/− mice also exhibited a shorter time to occlusion using the ferric-chloride injury model and reduced bleeding times compared with WT littermate controls. These results indicate that ELMO1 plays an important role in hemostasis and thrombosis in vivo. RhoG activity was enhanced in ELMO1−/− murine platelets compared with WT littermate controls in response to GPVI agonist. Together, these data suggest that ELMO1 negatively regulates GPVI-mediated thrombus formation via RhoG.
Introduction
Platelets are anucleate cells that are crucial mediators of thrombosis and hemostasis. Under physiological conditions, platelets are maintained in a quiescent state within the vasculature. Upon exposure of the subendothelial collagen during vascular injury, the glycoprotein VI (GPVI)1 receptor initiates platelet activation. The interaction between GPVI and collagen results in platelet shape change, thromboxane A2 (TXA2) synthesis, and granular secretion, eventually leading to the activation of integrin αIIbβ3. TXA2 and adenosine 5′-diphosphate act in an autocrine and paracrine manner to further enhance platelet activation and recruit platelets to the site of injury. The activation of integrin αIIbβ3 leads to platelet aggregation and stabilization of the thrombus.
GPVI is constitutively associated with the Fc receptor-γ chain (FcRγ), which contains an immune tyrosine-based activation motif.1 The interaction of collagen to the GPVI/FcRγ complex leads to phosphorylation of the immune tyrosine-based activation motif by the constitutively associated Src-family tyrosine kinases Fyn and Lyn,2,3 initiating the activation of spleen tyrosine kinase (Syk).4-6 The activation of Syk leads to downstream signaling events, including the activation of class I phosphoinositide 3-kinases (PI3Ks).7 In platelets, the activation of class I PI3K is not restricted to the GPVI pathway but is also activated downstream of P2Y128 and αIIbβ3 receptors.8 The inhibition of class I PI3K with pharmacological and genetic approaches leads to diminished platelet functional responses.8-10 PI3K phosphorylates the 3-hydroxyl group of the inositol ring of phosphatidylinositol, leading to the generation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate in the plasma membrane.8 PIP3 formation in turn leads to the recruitment of pleckstrin homology (PH) domain–containing proteins to the plasma membrane11 such as Bruton tyrosine kinase (Btk)12-14 and Ras GTPase-activating protein 3 (RASA3).15
Engulfment and cell motility (ELMO) is a scaffold protein with no catalytic activity.16 There are 3 mammalian isoforms of ELMO proteins, ELMO 1-3. These proteins contain armadillo repeats at the N terminus and a PH domain followed by a proline-rich region at the C terminus.16 ELMO1 and ELMO2 are well known to regulate actin cytoskeletal rearrangement via Rac1 upon interaction with dedicator of cytokinesis (DOCK), a guanine nucleotide exchange factor.16 Active RhoG, a small GTPase, can also interact with ELMO1 and regulate the actin cytoskeleton via the ELMO, DOCK, and Rac1 axis, and it is involved in cell migration,17-19 engulfment of apoptotic cells,16 and neurite outgrowth.20 However, in platelets, active RhoG interacts with ELMO1 and DOCK180 but does not appear to regulate the actin cytoskeleton via Rac1 following GPVI-mediated platelet activation.21 ELMO1 expression has been established in human platelets,21 but the function of ELMO1 is not yet known.
In the present study, we screened for platelet proteins that interacted with PIP3 using a proteomic approach and found ELMO1. We used ELMO1−/− mice to characterize the functional role of ELMO1 in platelets. We provide evidence that absence of ELMO1 leads to enhanced GPVI-mediated platelet functional responses. ELMO1 also negatively regulates integrin αIIbβ3–mediated platelet spreading. Our data also demonstrate that ELMO1 negatively regulates hemostasis and thrombotic functions in vivo. Finally, we show that ELMO1 negatively regulates Syk activation in platelets downstream of the GPVI signaling pathway via RhoG.
Materials and methods
Materials
All materials were obtained from ThermoFisher Scientific unless noted. PI(3,4,5)P3 PIP Beads were obtained from Echelon (Salt Lake City, UT). 2-MeSADP, acetylsalicylic acid, apyrase (type V), and anti-ELMO1 antibody were obtained from Sigma (St. Louis, MO). Collagen-related peptide (CRP)–XL was purchased from Richard Farndale at the University of Cambridge. AYPGKF was obtained from GenScript (Piscataway, NJ). Type I collagen and CHRONO-LUME were obtained from CHRONO-LOG Corporation (Havertown, PA). Anti-Syk-01 and anti-PLCγ2 were obtained from Santa Cruz Biotechnology (Dallas, TX). Antibodies against β-actin, Rac1, phosphorylated Syk (Tyr525/6), PLCγ2 (Tyr759), PLCγ2 (Tyr1217), and Akt (Ser473) were purchased from Cell Signaling Technology (Danvers, MA). Anti-Akt was obtained from OriGene (Rockville, MD) Infrared dye–labeled goat anti-mouse and anti-rabbit antibodies were purchased from LI-COR (Lincoln, NE). Anti-RhoG antibody was purchased from Millipore (Temecula, CA). Anti– fluorescein isothiocyanate (FITC)-GPVI, anti-FITC-P-selectin, and anti-phycoerythrin-JON/A were purchased from Emfret (Würzburg, Germany). Prostaglandin E1 was purchased from Enzo (New York, NY). Rhodocytin was provided by J.A.E.
Preparation of human platelets
Blood was collected from informed healthy donors into one-sixth volume of ACD (85 mM sodium citrate, 71.4 mM citric acid, and 111 mM dextrose). Protocol approval was obtained from the institutional review board of Temple University and informed consent was provided prior to blood donation, in accordance with the Declaration of Helsinki. Platelets were isolated as previously described.22 The platelet count was adjusted to 2 × 108 cells/mL unless otherwise stated. For the PIP3 pull-down assay, platelets were washed with piperazine-N,N′-bis(2-ethanesulfonic acid) buffer (137 mM sodium chloride, 2.7 mM potassium chloride, 2 mM magnesium chloride, 0.42 mM sodium phosphate monobasic, 0.1% dextrose, 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) adjusted to pH 6.5) containing 0.2 U/mL apyrase, 500 μM EGTA, and 10 nM prostaglandin E1 (PGE1) prior to being resuspended in Tyrode buffer (137 mM sodium chloride, 2.7 mM potassium chloride, 2 mM magnesium chloride, 0.42 mM sodium phosphate monobasic, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 0.1% dextrose adjusted to pH 7.4). The platelet count was adjusted to 2 × 109 cells/mL for the PIP3 pull-down assay.
Preparation of murine platelets
All mice were maintained and housed in a specific-pathogen-free facility, and animal procedures were carried out in accordance with the institutional guidelines after the Temple University Institutional Animal Care and Use Committee approved the study protocol. ELMO1-deficient mice (ELMO1−/−) were obtained from Kodi Ravichandran at the University of Virginia School of Medicine.23 Age-matched wild-type (WT) littermates were used as controls. Whole blood count was measured using the Hemavet (Drew Scientific, Miami Lakes, FL). Whole blood was collected from 10- to 12-week-old mice via cardiac puncture into one-tenth volume of 3.8% sodium citrate, and platelets were isolated as previously described.22
Sample preparation
Reactions were stopped with one-tenth volume of 6.6 N perchloric acid to precipitate proteins unless otherwise stated. The samples were centrifuged at 15 000g for 5 minutes at 4°C and washed with water, and the proteins were solubilized in 1x sample buffer (0.1 M Tris-base, 1% glycerol, 2% sodium dodecyl sulfate [SDS], and 100 mM dithiothreitol). The samples were boiled at 95°C for 10 minutes. For the PIP3 pull-down assay, resting human platelets were lysed using an equal volume of 2x cold NP-40 buffer (150 mM sodium chloride, 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 2 mM EGTA, and 0.5% Nonidet p-40 adjusted to pH 7.4) containing Halt Protease and Phosphatase cocktail solution (Pierce, Rockford, IL) and centrifuged at 12 000g for 10 minutes at 4°C to pellet cell debris.
PIP3 pull-down assay
The PIP3 pull-down assay was performed as per the manufacturer’s instructions. Briefly, cleared lysates were incubated with 100 μL PI(3,4,5)P3 PIP beads or control beads overnight at 4°C. Beads were washed 3 times with 1x cold NP-40 buffer, eluted with 2x SDS-sample buffer (Boston BioProducts), and boiled at 95°C for 10 minutes.
In-gel trypsin digestion and LC-MS analysis
Samples from the PIP3 pull-down were subjected to SDS polyacrylamide gel electrophoresis (PAGE) and analyzed by liquid chromatography-mass spectroscopy (LC-MS) as previously described.24
Immunoblotting
Human and murine platelet lysates were subjected to SDS-PAGE and western blot analysis as previously described.22 Blots were analyzed using the Odyssey imaging system (LI-COR).
Platelet aggregation and dense granule secretion
Platelet aggregation and ATP secretion were simultaneously detected using a lumiaggregometer (CHRONO-LOG Corporation) at 37°C while stirring. ATP secretion was detected using CHRONO-LUME (CHRONO-LOG Corporation), a luciferin/luciferase reagent.
Flow cytometry
GPVI surface expression (anti-FITC-GPVI), integrin αIIbβ3 activation (anti-phycoerythrin-JON/A), and surface exposure of P-selectin (anti-FITC-P-selectin) in murine platelets were determined as previously described.22 The samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences). Appropriate isotype controls were used.
Thromboxane generation
Murine platelets were stimulated with varying concentrations of CRP or collagen for 4 minutes, and the generated thromboxane was determined as previously descrbed.25
Flow over collagen
The flow over collagen assay was performed as previously described.26 Briefly, whole blood was isolated from age-matched mice (10-12 weeks) by cardiac puncture, anticoagulated using PPACK (Enzo) and Heparin (Sigma), and perfused over a collagen-coated dish using a chamber from GlycoTech (Gaithersburg, MD) under arterial (1000 s−1) and venous (200 s−1) shear rates. Thrombus formation was observed using Nikon Eclipse TE300 inverted microscope (200× objective) and analyzed using ImageJ (National Institutes of Health) analysis.
Pulmonary thromboembolism
The pulmonary thromboembolism model was performed as previously described,27 with the following changes: age-matched mice (age 10-12 weeks) were weighed and anesthetized with ketamine/xylazine. Mice were then injected IV with 400 μg/kg collagen and 60 μg/kg epinephrine or phosphate-buffered saline as a control, and the time to cessation of respiration was recorded. Upon respiratory arrest, the heart was perfused with Chicago sky blue dye to confirm pulmonary emboli.
FeCl3 injury model
The left carotid artery was injured using ferric chloride (FeCl3) as previously described.28 Briefly, 10- to 12-week-old mice were anesthetized and the carotid artery was exposed to filter paper (1 × 1 mm) saturated with 7.5% FeCl3 for 90 seconds, and the flow rate was recorded using Transonic T402 flowmeter (Ithaca, NY). The time to occlusion was defined as the time required for the flow rate to reach 0 mL/min. The operator was blinded to mouse genotype while performing all experiments.
Tail bleeding
Four-week-old mice were anesthetized, the distal 3 mm of the tail was cut, and the tail was immediately immersed in 37°C saline. The time for bleeding to halt was recorded. Bleeding was manually stopped when mice bled for >10 minutes.
Platelet spreading
Washed murine platelets (1 × 107 cells/mL) were platelets onto fibrinogen-coated plates, and the platelets were fixed with formalin for 10 minutes at the indicated times. Platelets were permeabilized with 0.1% Triton X-100 and stained with FITC-phalloidin. Images were obtained using Leica DM IRE with a TCS SL confocal system using Leica Imaging software.
Expression of ELMO1-GST and the measurement of RhoG activation
The expression of bacterially expressed ELMO1 glutathione S-transferase (GST) and the measurement of RhoG activity were determined as previously described.29
Statistics
All statistical analyses were performed using KaliedaGraph and Microsoft Excel. Data were represented as mean ± standard error (SE) of at least 3 independent experiments. All statistics were analyzed using a Student t test. P < .05 was considered statistically significant.
Results
ELMO1 is associated with PIP3 in platelets
Class I PI3K is an important signaling molecule that is activated downstream of various receptors in platelets.8 The activation of class I PI3K leads to the generation of PIP3 and, in turn, the recruitment of PH-domain–containing proteins to the plasma membrane.8 In platelets, RASA3 and Btk are known to interact with PIP330 and are important mediators of platelet activation.31,32 However, whether there are other proteins that can interact with PIP3 in platelets is not known. Therefore, we used a proteomic approach to identify novel PIP3-interacting proteins in resting human platelets by performing a pull-down assay using PIP3 beads. We used control beads to detect nonspecific interactions. Proteins bound to PIP3 or control beads were separated by SDS-PAGE and identified by LC-MS. When compared with control beads, 24 proteins out of 154 proteins bound specifically with PIP3 (supplemental Table 1). Within these 24 proteins, we found 5 that are known to contain PH domains (Table 1). It is well known that RASA3,15 Btk,13,14 cytohesin-2,33,34 and dual adapter for phosphotyrosine and 3-phosphotyrosine and 3-phosphoinositide (DAPP1)35,36 can interact with PIP3 in nucleated cells. However, whether or not ELMO1 can associate with PIP3 in platelets has not been previously established. Here, we show that ELMO1 associates with PIP3 beads in platelets by western blot analysis, and we used Btk as a positive control, since Btk has been previously shown to interact with PIP3 in platelets30 (Figure 1A). This indicates that endogenous ELMO1 associates with PIP3 or a protein that interacts with PIP3 in platelets.
Summary of PH-domain–containing proteins from the proteomics data
| Accession no. | Protein | Score | Coverage | No. of peptides |
|---|---|---|---|---|
| Q14644 | RASA3 | 2029.17 | 27.22 | 24 |
| Q06187 | Tyrosine-protein kinase Btk | 473.89 | 25.49 | 14 |
| Q99418 | Cytohesin-2 | 120.69 | 11.50 | 4 |
| Q9UN19 | DAPP1 | 49.84 | 10.36 | 2 |
| Q92556 | ELMO1 | 41.93 | 1.51 | 1 |
| Accession no. | Protein | Score | Coverage | No. of peptides |
|---|---|---|---|---|
| Q14644 | RASA3 | 2029.17 | 27.22 | 24 |
| Q06187 | Tyrosine-protein kinase Btk | 473.89 | 25.49 | 14 |
| Q99418 | Cytohesin-2 | 120.69 | 11.50 | 4 |
| Q9UN19 | DAPP1 | 49.84 | 10.36 | 2 |
| Q92556 | ELMO1 | 41.93 | 1.51 | 1 |

ELMO1 associates with PIP3in platelets. (A) Resting human platelets (1 × 109 cells/mL) were lysed with NP-40 lysis buffer and incubated with PI(3,4,5)P3 PIP beads or control beads overnight at 4°C. Pull-downs were analyzed by western blot and probed with ELMO1 or Btk antibody. Blots are representative of at least 3 independent experiments. (B) The proteins from resting washed human, WT littermate control, and ELMO1−/− murine platelets were precipitated and analyzed by western blot. The blots were probed for ELMO1 and β-actin as a loading control. Blots are representative of at least 3 independent experiments. MW, molecular weight marker.
ELMO1 associates with PIP3in platelets. (A) Resting human platelets (1 × 109 cells/mL) were lysed with NP-40 lysis buffer and incubated with PI(3,4,5)P3 PIP beads or control beads overnight at 4°C. Pull-downs were analyzed by western blot and probed with ELMO1 or Btk antibody. Blots are representative of at least 3 independent experiments. (B) The proteins from resting washed human, WT littermate control, and ELMO1−/− murine platelets were precipitated and analyzed by western blot. The blots were probed for ELMO1 and β-actin as a loading control. Blots are representative of at least 3 independent experiments. MW, molecular weight marker.
ELMO1 negatively regulates platelet aggregation and granule secretion downstream of GPVI
We used ELMO1−/− mice23 to study the function of ELMO1 in platelets. As previously reported, ELMO1 is present in human platelets,21 and we also found ELMO1 present in murine platelets (Figure 1B). Furthermore, the absence of ELMO1 was confirmed in ELMO1−/− platelets (Figure 1B).
To investigate the function of ELMO1 in platelets, we performed aggregation and dense granule secretion experiments using platelets from ELMO1−/− and WT littermate control mice. In response to AYPGKF (PAR4 agonist) and 50 nM 2-MeSADP (data not shown), platelet aggregation and dense granule secretion were unaltered in platelets isolated from ELMO1−/− mice compared with WT littermate controls (Figure 2A-D). However, CRP (GPVI agonist)-induced platelet aggregation and dense granule secretion were enhanced in ELMO1−/− platelets in a concentration-dependent manner (Figure 2A-D). Similarly, dense granule secretion in response to low doses of the physiological GPVI agonist collagen was also potentiated in ELMO1−/− platelets (Figure 2A-D). Both P-selectin exposure and integrin αIIbβ3 activation are also enhanced in the ELMO1−/− platelets compared with the WT littermate control platelets in response to the GPVI agonist CRP (Figure 2E-F). This indicates that ELMO1 negatively regulates platelet aggregation, granule secretion, and integrin αIIbβ3 activation specifically downstream of the GPVI pathway.

Enhanced aggregation and granular secretion following GPVI stimulation in ELMO1−/−platelets. Representative aggregation (A) and dense granule secretion tracings (B) of washed platelets from ELMO1−/− or WT littermate controls activated with the indicated agonists for 4 minutes. Platelet aggregation and dense granule secretion was detected by lumiaggregometry under stirring conditions at 37°C. Quantification of extent of aggregation (C) and dense granule secretion (D) of at least 3 independent experiments from panels A and B, respectively. Data are presented as mean ± SE and were analyzed by Student t test (*P < .05). Flow cytometry analysis of P-selectin exposure (E) and activated integrin αIIbβ3 (F) of washed murine platelets from ELMO1−/− or WT littermate controls activated with the indicated concentrations of CRP for 10 minutes at 37°C. Data are presented as fold increase over basal ± SE from at least 3 independent experiments and were analyzed by Student t test (*P < .05). A.U, arbitrary units.
Enhanced aggregation and granular secretion following GPVI stimulation in ELMO1−/−platelets. Representative aggregation (A) and dense granule secretion tracings (B) of washed platelets from ELMO1−/− or WT littermate controls activated with the indicated agonists for 4 minutes. Platelet aggregation and dense granule secretion was detected by lumiaggregometry under stirring conditions at 37°C. Quantification of extent of aggregation (C) and dense granule secretion (D) of at least 3 independent experiments from panels A and B, respectively. Data are presented as mean ± SE and were analyzed by Student t test (*P < .05). Flow cytometry analysis of P-selectin exposure (E) and activated integrin αIIbβ3 (F) of washed murine platelets from ELMO1−/− or WT littermate controls activated with the indicated concentrations of CRP for 10 minutes at 37°C. Data are presented as fold increase over basal ± SE from at least 3 independent experiments and were analyzed by Student t test (*P < .05). A.U, arbitrary units.
Enhanced GPVI-mediated thromboxane generation in the ELMO1−/− platelets
We investigated the contribution of ELMO1 in thromboxane generation downstream of the GPVI pathway. In response to either CRP or collagen, thromboxane generation was enhanced in the ELMO1−/− platelets compared with the WT littermate control platelets in a concentration-dependent manner (Figure 3A). In order to determine whether or not feedback contributes to the enhancement of GPVI-mediated platelet aggregation and secretion observed in the ELMO1−/− platelets, we performed aggregation and secretion experiments in the presence of 10 μM indomethacin, 10 μM MRS-2179, and 100 nM AR-C69931MX. Both platelet aggregation and dense granule secretion were enhanced in the ELMO1−/− platelets compared with WT littermate controls in response to the GPVI agonist CRP (Figure 3B-C). This indicates that ELMO1 negatively regulates GPVI-mediated platelet functional responses independent of feedback.

Enhanced thromboxane generation in response to GPVI agonists in ELMO1−/−platelets. (A) Washed murine platelets were activated with the indicated concentrations of CRP or collagen for 4 minutes, and the reaction was stopped via flash-freezing. Thromboxane B2 generation was determined using an ELISA kit as per the manufacturer’s instructions. Data are presented as mean ± SE of at least 3 independent experiments and were analyzed by Student t test (*P < .05). (B) Representative aggregation tracings and dense granule secretion of washed platelets from ELMO1−/− or WT littermate controls preincubated with the feedback inhibitors (FBI’s) 10 μM indomethacin, 10 μM MRS-2179, and 100 nM AR-C69931MX for 5 minutes and activated with indicated concentration of CRP for 3 minutes. (C) Quantitation of aggregation and ATP secretion showing the mean ± standard error of the mean and analyzed by Student t test (*P < .05).
Enhanced thromboxane generation in response to GPVI agonists in ELMO1−/−platelets. (A) Washed murine platelets were activated with the indicated concentrations of CRP or collagen for 4 minutes, and the reaction was stopped via flash-freezing. Thromboxane B2 generation was determined using an ELISA kit as per the manufacturer’s instructions. Data are presented as mean ± SE of at least 3 independent experiments and were analyzed by Student t test (*P < .05). (B) Representative aggregation tracings and dense granule secretion of washed platelets from ELMO1−/− or WT littermate controls preincubated with the feedback inhibitors (FBI’s) 10 μM indomethacin, 10 μM MRS-2179, and 100 nM AR-C69931MX for 5 minutes and activated with indicated concentration of CRP for 3 minutes. (C) Quantitation of aggregation and ATP secretion showing the mean ± standard error of the mean and analyzed by Student t test (*P < .05).
ELMO1 inhibits platelet spreading
ELMO1 has been implicated in integrin-mediated spreading and migration in nucleated cells.17,20 Hence, we wanted to determine whether ELMO1 contributes to integrin αIIbβ3-mediated outside-in signaling by evaluating platelet adhesion and spreading over immobilized fibrinogen. Both littermate control and ELMO1−/− platelets adhered to immobilized fibrinogen (Figure 4). However, the kinetics of filopodia formation was enhanced in the ELMO1−/− platelets compared with the WT littermate control. This indicates that ELMO1 negatively regulates platelet spreading.

ELMO1 regulates integrin-mediated platelet spreading. (A) Representative images of murine platelets incubated on fibrinogen-coated plates for the times indicated and fixed and stained with FITC-phalloidin. The images were obtained using confocal microscope. Scale bars, 5 μM. (B) Quantification of platelets with a filopodial phenotype expressed as a percentage of adhered platelets. At least 3 fields containing at least 25 platelets were analyzed per replicate (at least 2 replicates per experiment). Data were analyzed via Student t test (*P < .05). WT and ELMO1−/−, n = 4.
ELMO1 regulates integrin-mediated platelet spreading. (A) Representative images of murine platelets incubated on fibrinogen-coated plates for the times indicated and fixed and stained with FITC-phalloidin. The images were obtained using confocal microscope. Scale bars, 5 μM. (B) Quantification of platelets with a filopodial phenotype expressed as a percentage of adhered platelets. At least 3 fields containing at least 25 platelets were analyzed per replicate (at least 2 replicates per experiment). Data were analyzed via Student t test (*P < .05). WT and ELMO1−/−, n = 4.
ELMO1 regulates thrombus formation in vitro
Our previous data suggest that ELMO1 specifically regulates GPVI-mediated platelet activation. Therefore, we investigated the role of ELMO1 in thrombus formation in an in vitro flow over collagen model under arterial (1000 s−1) and venous (200 s−1) shear conditions. Whole blood from ELMO1−/− mice exhibited enhanced thrombus formation compared with the WT littermate control in both arterial and venous conditions (Figure 5). This is consistent with the enhanced dense granule secretion and thromboxane generation observed with ex vivo ELMO1−/− murine platelets in response to GPVI agonists.

ELMO1 regulates thrombus formation in vitro. Representative light microscopic image of thrombus formation on a collagen-coated surface (A) and percent thrombus area (B). Whole blood from WT littermate controls and ELMO1−/− mice was perfused over a collagen (50 μg/mL)–coated surface at arterial shear rate 1000 s−1 or venous shear rate 200 s−1 for 4 minutes. The direction of flow was right to left. (B) Images were analyzed using ImageJ, and data are presented as percent thrombus area ± SE. Data were analyzed by Student t test (*P < .05). WT and ELMO1−/−, n = 3.
ELMO1 regulates thrombus formation in vitro. Representative light microscopic image of thrombus formation on a collagen-coated surface (A) and percent thrombus area (B). Whole blood from WT littermate controls and ELMO1−/− mice was perfused over a collagen (50 μg/mL)–coated surface at arterial shear rate 1000 s−1 or venous shear rate 200 s−1 for 4 minutes. The direction of flow was right to left. (B) Images were analyzed using ImageJ, and data are presented as percent thrombus area ± SE. Data were analyzed by Student t test (*P < .05). WT and ELMO1−/−, n = 3.
ELMO1 regulates both thrombosis and hemostasis in vivo
To determine if ELMO1 plays a similar role in vivo, we used a pulmonary thromboembolism model via IV administration of collagen and epinephrine in WT littermate and ELMO1−/− mice. ELMO1−/− mice exhibited a lower survival rate than WT littermate controls (Figure 6A). We also evaluated the functional implication of the absence of ELMO1 on arterial thrombus formation upon FeCl3 injury. ELMO1−/− mice exhibited shorter times to occlusion than WT littermate controls (Figure 6B). This indicates that ELMO1 negatively regulates thrombus formation in vivo.
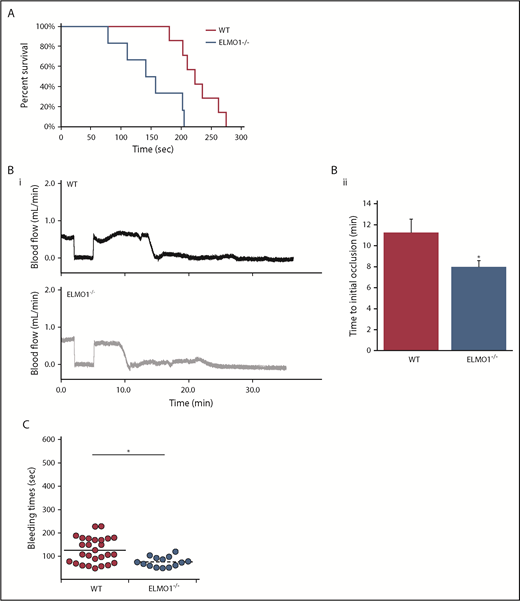
ELMO1 regulates thrombus formation and hemostasis in vivo. (A) Survival curves of WT and ELMO1−/− mice in a pulmonary thromboembolism model. Time to cessation of respiration was recorded after IV administration of 400 μg/kg collagen and 60 μg/kg epinephrine or phosphate-buffered saline. WT, n = 7; ELMO1−/−, n = 6. (B) Representative carotid artery blood flow tracings (i) and average time to occlusion (ii) from WT and ELMO1−/− mice. A carotid artery was isolated from the indicated mice and was exposed to 7.5% FeCl3 injury for 90 seconds. Data are presented as mean ± SE. WT, n = 12; ELMO1−/−, n = 14. Statistical analysis was performed using a Mann-Whitney U test (*P < .05). (C) Tail bleeding times of WT and ELMO1−/− mice. The distal 3 mm of the tail was cut, and the tail was immersed in 37°C saline. The time it took for bleeding to stop was recorded. WT, n = 38; ELMO1−/−, n = 18. Statistical analysis was performed using a Student t test (*P < .05).
ELMO1 regulates thrombus formation and hemostasis in vivo. (A) Survival curves of WT and ELMO1−/− mice in a pulmonary thromboembolism model. Time to cessation of respiration was recorded after IV administration of 400 μg/kg collagen and 60 μg/kg epinephrine or phosphate-buffered saline. WT, n = 7; ELMO1−/−, n = 6. (B) Representative carotid artery blood flow tracings (i) and average time to occlusion (ii) from WT and ELMO1−/− mice. A carotid artery was isolated from the indicated mice and was exposed to 7.5% FeCl3 injury for 90 seconds. Data are presented as mean ± SE. WT, n = 12; ELMO1−/−, n = 14. Statistical analysis was performed using a Mann-Whitney U test (*P < .05). (C) Tail bleeding times of WT and ELMO1−/− mice. The distal 3 mm of the tail was cut, and the tail was immersed in 37°C saline. The time it took for bleeding to stop was recorded. WT, n = 38; ELMO1−/−, n = 18. Statistical analysis was performed using a Student t test (*P < .05).
To investigate whether ELMO1 has any hemostatic function, we performed a tail bleeding assay using ELMO1−/− and WT littermate mice. ELMO1−/− mice have shorter bleeding times than WT littermate controls, indicating the importance of ELMO1 in hemostasis (Figure 6C). Hematological parameters, including platelet count and mean platelet volume, were unaltered in ELMO1−/− mice compared with WT littermate controls (supplemental Table 2). This indicates the importance of ELMO1 in both hemostasis and thrombosis.
ELMO1 negatively regulates Syk downstream of GPVI
In nucleated cells, ELMO1 regulates the actin cytoskeleton via Rac1 upon interaction with DOCK proteins.16,37 It was previously reported that Rac1 regulates GPVI-mediated platelet responses, but not PAR-mediated platelet responses.38 Hence, we evaluated Rac1 activity in ELMO1−/− platelets downstream of GPVI signaling. Rac1 activity is enhanced in ELMO1−/− platelets compared with WT controls (Figure 7A), indicating that ELMO1 regulates Rac1 activity in platelets.

ELMO1 regulates RhoG activity in the GPVI pathway. (A) Rac1 activation in response to CRP in the absence of ELMO1. Washed platelets were stimulated with 1.5 μg/mL CRP for the indicated times and lysed with NP-40 lysis buffer, and active Rac was pulled down using GST-PAK-RBD. Active Rac1 was detected via western blot analysis using a specific antibody to Rac1. (B-C) Time course of ELMO1−/− and WT platelets activated with 1.25 μg/mL CRP at the indicated time points under stirring conditions at 37°C. Proteins were precipitated and analyzed by western blot using indicated antibodies. (D) Surface expression of GPVI in washed murine platelets from WT and ELMO1−/− mice assessed by flow cytometry. GPVI surface expression is a representative of at least 3 independent experiments. (E) RhoG activity in ELMO1−/− and WT murine platelets in response to CRP. Washed murine platelets were activated with 10 μg/mL CRP for 1 minute and lysed with NP-40 lysis buffer. Active RhoG was pulled down using GST-ELMO1 fusion protein, and RhoG activity was detected by western blot. (F) Model of GPVI signaling by ELMO1 in platelets. All western blot images are representative of at least 3 independent experiments.
ELMO1 regulates RhoG activity in the GPVI pathway. (A) Rac1 activation in response to CRP in the absence of ELMO1. Washed platelets were stimulated with 1.5 μg/mL CRP for the indicated times and lysed with NP-40 lysis buffer, and active Rac was pulled down using GST-PAK-RBD. Active Rac1 was detected via western blot analysis using a specific antibody to Rac1. (B-C) Time course of ELMO1−/− and WT platelets activated with 1.25 μg/mL CRP at the indicated time points under stirring conditions at 37°C. Proteins were precipitated and analyzed by western blot using indicated antibodies. (D) Surface expression of GPVI in washed murine platelets from WT and ELMO1−/− mice assessed by flow cytometry. GPVI surface expression is a representative of at least 3 independent experiments. (E) RhoG activity in ELMO1−/− and WT murine platelets in response to CRP. Washed murine platelets were activated with 10 μg/mL CRP for 1 minute and lysed with NP-40 lysis buffer. Active RhoG was pulled down using GST-ELMO1 fusion protein, and RhoG activity was detected by western blot. (F) Model of GPVI signaling by ELMO1 in platelets. All western blot images are representative of at least 3 independent experiments.
The activation of Syk4-6 via the GPVI/FcRγ complex leads to downstream signaling events leading to activation of Rac1,39,40 PI3K,7 and PLCγ2.41 Thus, we evaluated Syk phosphorylation downstream of GPVI signaling in ELMO1−/− platelets to determine whether altered Rac1 activation is a result of altered Syk phosphorylation. The phosphorylation of Syk is enhanced in as early as 30 seconds in ELMO1−/− platelets compared with WT littermate control platelets in response to CRP (Figure 7B). Furthermore, both PLCγ2 (Figure 7B) and Akt phosphorylation (Figure 7C), a measure of PI3K activity, are also enhanced in ELMO1−/− platelets downstream of GPVI signaling. This indicates that ELMO1 regulates Syk downstream of the GPVI signaling pathway and that the enhanced Rac1 activity in ELMO1−/− platelets is due to enhanced Syk phosphorylation. Notably, GPVI surface expression is unaltered with ELMO1 deficiency (Figure 7D). This indicates that ELMO1 negatively regulates Syk within the GPVI signaling pathway.
ELMO1 regulates RhoG activity in the GPVI signaling pathway
We and others have previously shown that RhoG regulates GPVI-mediated platelet activation and thrombus formation.21,29 RhoG was also activated as early as 30 seconds in response to CRP and it regulates GPVI-mediated platelet activation by regulating Syk.29 Active RhoG also interacts with ELMO1 in platelets,21 but the functional implication of this interaction is not understood. Therefore, we used full-length bacterially expressed and purified ELMO1-GST as bait for active RhoG in platelets from ELMO1−/− and WT littermate controls in response to CRP. RhoG activity was enhanced in the ELMO1−/− platelets in response to CRP compared with WT littermate controls (Figure 7E). This indicates that ELMO1 regulates RhoG activity leading to GPVI-mediated platelet activation.
We previously reported that CLEC-2–mediated platelet activity was unaltered in RhoG-deficient platelets.29 To provide further evidence that ELMO1 exerts its influence via RhoG, we stimulated washed WT, ELMO1−/−, and RhoG−/− platelets with the CLEC-2 agonist rhodocytin and measured aggregation and secretion. There was no difference in platelet reactivity to rhodocytin regardless of genotype, with the exception of both RhoG+/+ and RhoG−/− platelets at 5 nM rhodocytin (Figure 8A). At this threshold concentration neither the RhoG knockout platelets nor the littermate control platelets reacted, while the ELMO knockout and littermate control platelets did. This is likely due to strain differences, as the ELMO mice are on a C57/bl6 background, while the RhoG mice are on a mixed background. However, at 10 nM rhodocytin there is no difference observed regardless of genotype (Figure 8B). Therefore, we believe that, similar to RhoG, ELMO1 is dispensable for CLEC-2-mediated platelet reactivity.

ELMO1 deficiency is dispensable for CLEC-2–mediated platelet reactivity. Washed platelets isolated from WT, ELMO1−/−, and RhoG−/− mice were diluted to 1.5 × 108 and stimulated with either 5 nM (A) or 10 nM (C) rhodocytin, and subsequent aggregations and secretions were recorded. (B,D) Quantification of ATP secretion expressed as mean ± standard error of the mean following platelet stimulation with either 5 nM or 10 nM rhodocytin.
ELMO1 deficiency is dispensable for CLEC-2–mediated platelet reactivity. Washed platelets isolated from WT, ELMO1−/−, and RhoG−/− mice were diluted to 1.5 × 108 and stimulated with either 5 nM (A) or 10 nM (C) rhodocytin, and subsequent aggregations and secretions were recorded. (B,D) Quantification of ATP secretion expressed as mean ± standard error of the mean following platelet stimulation with either 5 nM or 10 nM rhodocytin.
Discussion
The generation of PIP3 upon class I PI3K activation acts as a central hub to recruit downstream effectors such as Btk and RASA3, which are important mediators of platelet activation.8,12,31 However, there may be other PIP3-interacting proteins in platelets. Therefore, we performed a proteomic screen using PIP3 beads as bait to identify novel PIP3-interacting proteins. Our data agree with a previous study that identified PIP3-interacting proteins in platelets.42 We focused on PH-domain–containing proteins and found the following: RASA3, Btk, cytohesin-2, DAPP1, and ELMO1. Among these proteins, we studied ELMO1, since its function is unknown in platelets.
Using ELMO1−/− mice, we provide evidence that ELMO1 is important for platelet function and thrombosis. ELMO1−/− platelets exhibit enhanced platelet aggregation, granule secretion, and thromboxane generation specifically downstream of the GPVI pathway despite normal surface expression of GPVI. Consequently, whole blood from ELMO1−/− mice exhibits enhanced thrombus formation in an in vitro flow over collagen model. Consistently, we provide in vivo evidence of the importance of ELMO1 in thrombosis and hemostasis. However, ELMO1 is ubiquitously expressed, and the ELMO1−/− mice used in this study are a global knockout.23 Therefore, we cannot rule out the contribution of other cell types in thrombus formation in vivo. A platelet-specific conditional ELMO1−/− mouse would be required to determine ELMO1’s role in thrombus formation.
Additionally, this study provides evidence that ELMO1 negatively regulates integrin-mediated platelet spreading. This is consistent with previous studies in nucleated cells indicating ELMO1’s role in integrin-mediated spreading.17,20 How ELMO1 regulates platelet spreading remains to be understood. In nucleated cells, ELMO1 regulates integrin-mediated spreading via Rac1.17,20 In platelets, Rac1 is known to be involved in lamellipodia formation.38 Since we show that the kinetics of filopodia formation is enhanced in ELMO1−/− mice, we speculate that ELMO1’s function in integrin-mediated spreading is not via Rac1. Future studies are required to understand how ELMO1 regulates integrin-mediated platelet spreading.
PH-domain–containing proteins are known to interact with PIP3 in nucleated cells,11 and ELMO1-3 contain PH domain at the C terminus.16 A previous in vitro study revealed that the PH domain of ELMO1 does not interact with PIP3.43 However, in this study, we provide evidence that endogenous ELMO1 can interact with PIP3 in platelets. This could be due to ELMO1 indirectly interacting with PIP3 by associating with proteins such as DOCK in an endogenous system. DOCK proteins contain a DOCK homology region 1 domain that is well known to interact with PIP3 in nucleated cells,44-46 and ELMO1 can interact with DOCKA (DOCK180, 2, and 5) and DOCKB subfamilies (DOCK3 and 4).47 The proteomic screen in this study also shows DOCK5 as a PIP3-interacting protein in platelets. Therefore, it is possible that ELMO1 interacts with PIP3 indirectly via DOCK5. Future studies using ELMO1−/− and DOCK5−/− platelets may clarify whether endogenous ELMO1 directly or indirectly interacts with PIP3 in platelets.
Interestingly, we did not find ELMO2 and ELMO3 in our proteomic screen. However, by western blot analysis, we did find ELMO2, but not ELMO3, in human and murine platelets (data not shown), which suggests that ELMO2 may not be interacting with PIP3 in platelets. Furthermore, ELMO2 is upregulated (data not shown) in platelets from ELMO1−/− mice. However, ELMO2 does not seem to be compensating for the function of ELMO1, since the platelet functional responses are intact. The function of macrophages and fibroblasts isolated from ELMO1−/− mice was unaltered due to upregulation of ELMO2, suggesting a compensatory role for ELMO2 in these cells.23
Previous studies have shown that class I PI3K7,32 and Rac139,40 are activated downstream of Syk in the GPVI signaling pathway. In this study, we provide evidence that Syk phosphorylation and the signaling downstream of Syk, such as Rac1 activity, PLCγ2, and Akt phosphorylation, are enhanced in ELMO1−/− platelets downstream of GPVI. Therefore, we speculate that neither the ELMO1/PIP3 interaction nor Rac1 contributes to the GPVI-mediated platelet functional responses observed in the ELMO1−/− mice. Hence, we propose that the enhanced GPVI-mediated platelet responses observed in ELMO1−/− platelets are due to altered Syk phosphorylation.
How ELMO1 regulates Syk downstream of the GPVI pathway is not understood. We and others have previously shown that RhoG positively regulates GPVI-mediated platelet activation.21,29 Furthermore, we reported diminished Syk phosphorylation in RhoG-deficient platelets downstream of GPVI signaling. ELMO1 also specifically interacts with active RhoG in platelets.21 Therefore, we used recombinant ELMO1 as bait to detect active RhoG in the WT vs ELMO1−/− platelets downstream of GPVI signaling. Our data provide evidence that RhoG activity is enhanced in the ELMO1−/− murine platelets compared with WT platelets upon stimulation with CRP, indicating that ELMO1’s ability to regulate Syk may be due to its interaction with active RhoG. Therefore, we propose that upon binding to RhoG, ELMO1 inhibits RhoG activity downstream of GPVI.
The enhanced RhoG activity observed in ELMO1−/− platelets using recombinant ELMO1 as bait may be due to the absence of competition by endogenous ELMO1/active RhoG in ELMO1−/− platelets. Since the bait is added in excess to outcompete the endogenous ELMO1’s ability to bind active RhoG, we speculate that endogenous ELMO1 binding to active RhoG in WT platelets would be minimal. Currently, the only way to measure RhoG activity is by using recombinant ELMO1.
The ability of ELMO1 to inhibit RhoG activity relies on RhoG activation to occur first, since ELMO1 specifically interacts with active RhoG in platelets.21 This is consistent with previous studies in nucleated cells, where RhoG is known to be upstream of ELMO1.20 Therefore, we propose the following model (outlined in Figure 7E), which is supported by our CLEC-2 data, regarding the regulation of ELMO1 in platelets. Under physiological conditions, RhoG is maintained inactive. Upon vascular injury, activation of the GPVI/FcRγ complex leads to activation of RhoG.21,29 Activation of RhoG leads to phosphorylation of Syk, which in turn leads to activation of downstream signaling molecules such as PI3K and PLCγ2, eventually leading to platelet activation.29 Concomitantly to RhoG activation, ELMO1 interacts with and inhibits active RhoG21 to ensure platelets remain quiescent.
In conclusion, ELMO1 negatively regulates GPVI-mediated platelet activation and thrombus formation through binding to RhoG. ELMO1 also negatively regulates integrin-mediated platelet spreading.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Kodi Ravichandran (University of Virginia) for providing the ELMO1-deficient mice.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL132171, HL93231, HL137207, and HL137721 (S. Kunapuli) and American Heart Association grants 17SDG33370020 (J.K.) and 17SDG33350075 (J.E.A.).
Authorship
Contribution: A.P. designed and performed experiments, analyzed and interpreted data, and drafted the manuscript; J.K. designed and performed experiments, analyzed and interpreted data, and edited the manuscript; R.B. and D.B. performed experiments; J.E.A. and C.D. designed and performed experiments and analyzed and interpreted data; S. Kim collected data, provided direction, and analyzed data; J.A.E. designed experiments and analyzed data; S.M. performed proteomics for PIP3-interacting proteins; and L.G. and S. Kunapuli designed experiments and analyzed and interpreted data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Satya Kunapuli, Sol Sherry Thrombosis Research Center, Temple University, Room 414 MRB, 3420 N Broad St, Philadelphia, PA 19140; e-mail: spk@temple.edu.

