

Key Points
Ecto+ ILC neutralize extracellular ATP and suppress autologous T cells via the production of adenosine.
Human acute GVHD is associated with a depletion of tissue ecto+ ILC3 and a reduction in serum adenosine levels.

Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is often associated with chemotherapy- and radiotherapy-induced host tissue damage, leading to graft-versus-host disease (GVHD). Innate lymphoid cells (ILC) have an essential role in tissue homeostasis and tissue repair via their production of interleukin (IL)-22, which acts on intestinal stem cells. The tissue healing capacities of ILC via IL-22 in the context of allo-HSCT and GVHD has previously been demonstrated in a mouse model for acute GVHD. We investigated potential other ways of ILC-mediated tissue protection against GVHD. Tissue injury leads to the release of danger-associated molecular patterns (DAMPs). DAMPs interact with purinergic receptors and ectoenzymes on immune cells and induce pleiotropic effects, including activation of proinflammatory antigen-presenting cells and immunosuppressive effects via the generation of adenosine. Here, we report a novel subset of human ILC3 that coexpress the ectoenzymes CD39 and CD73 (ecto+ ILC3). Ecto+ ILC3 express RORγt and were present in the oral-gastrointestinal tract and bone marrow. ILC3 ectoenzyme expression is modulated by the proinflammatory cytokine IL-1β. Extracellular adenosine triphosphate (eATP) stimulated ecto+ ILC3 to produce IL-22 and adenosine. Activated ecto+ ILC3 suppressed autologous T-cell proliferation in coculture experiments via the production of adenosine. In allo-HSCT recipients, intestinal GVHD was associated with reduced proportions of ecto+ ILC3 and decreased levels of adenosine and its metabolite inosine. Taken together, ecto+ ILC3 have immunosuppressive properties, but in patients with GVHD, ecto+ ILC3 are depleted. A lack of ecto+ ILC3 and subsequent reduced capacity to neutralize DAMPs may contribute to the development of GVHD.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is a powerful tool to cure patients with malignant and nonmalignant hematologic diseases, but graft-versus-host responses, induced by the donor immune system, are a predominant cause of morbidity and mortality. Graft-versus-host disease (GVHD) results from conditioning regimen-related tissue damage of epithelial barriers that leads to the release of proinflammatory cytokines (such as interleukin 1β [IL-1β], tumor necrosis factor α [TNF-α]), and danger signals, such as danger-associated molecular patterns (DAMPs).
DAMPs, including the purine metabolites adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (NAD+) can induce inflammation by activation of antigen-presenting cells, but can also be metabolized into immune dampening derivatives such as adenosine.1 Extracellular ATP (eATP) and NAD+ can be hydrolyzed into adenosine by cell surface enzymes called ectoenzymes or ectonucleotidases. Ectoenzymes, such as CD38, CD39, and CD73, serve to bridge the communication between cells and their microenvironment. Extracellular ATP and extracellular adenosine diphosphate (eADP) are phosphohydrolyzed by CD39 into adenosine monophosphate (eAMP) and then into adenosine via CD73.2 An alternative mechanism that drives the generation of extracellular adenosine is the conversion of NAD+ into eAMP and subsequently into adenosine via a chain reaction mediated by CD38 along with the ectonucleotide pyrophosphatase CD203a (or PC-1) and CD73. In a final step, adenosine is inactivated and degraded into its metabolite inosine by the surface molecule dipeptyl-peptidase CD26 in complex with adenosine deaminase (ADA).3,4 Adenosine and inosine exert direct and indirect immunosuppressive effects.4 Adenosine, CD39, and CD73 have been shown to limit the severity of GVHD and enhance graft-versus-leukemia effects in experimental models.5,6 For instance, CD39 activity on murine T regulatory (Treg) cells is linked to protective functions by inhibiting Notch1-signaling by conventional T cells.7
Conversely, eATP and eNAD+ can also have immunostimulatory effects when sensed by purinergic receptors such as P1 receptors (A1, A2A, A2B, and A3), P2X receptors (P2X1-7 subtypes), or P2Y receptors (P2Y1,2,4,6,11-14 subtypes). In the context of GVHD, eATP mediates inflammatory responses via the purine P2 receptor P2X7R, which has been associated with expansion of allogeneic donor T cells and suppression of Treg cells, causing progression of GVHD.8 In line with this, a broad-spectrum of P2X7 antagonists attenuated murine GVHD without interfering with the graft-versus-leukemia response.9 In addition, the selective A2A adenosine receptor agonist ATL146 has been reported to decrease the severity of murine GVHD by enhancing the number of donor-derived Treg cells.10
Innate lymphoid cells (ILC) are tissue-resident lymphocytes that are important in the innate effector response against pathogenic microbes, help to maintain epithelial integrity at barrier surfaces, and are important in tissue homeostasis and repair. At this time, 5 populations of ILC have been identified; namely, natural killer (NK) cells, ILC1, ILC2, ILC3, and lymphoid tissue inducer cells.11 In the intestine, ILC3 are strategically positioned within the intestinal lamina propria, controlling the homeostasis by rapidly responding to environmental cues and interacting with other immune and nonimmune cells. Importantly, ILC3 contribute to the integrity of the epithelial barrier by inducing mucus production, antimicrobial peptide production, and fucosylation via the production of IL-22 in response to dietary retinoic acid (RA) and aryl hydrocarbon receptor ligands.12-14 Regulatory ILC, producing IL-10, have been detected in human intestines15 and in nasal tissues from patients with chronic rhinosinusitis.16
ILC also can contribute to tissue protection in the context of GVHD. In a murine allo-HSCT model, it has been demonstrated that IL-22-producing ILC3 enhanced intestinal stem cell functions.17 In addition, co-transfusion of ILC with T cells led to the prevention of murine GVHD.18 In humans, we have shown that ILC are depleted from the blood of patients who undergo conditioning therapy before allo-HSCT. Interestingly, patients with a relatively rapid recovery of ILC numbers after induction chemotherapy, before allo-HSCT, experienced less mucositis and less acute GVHD after allo-HSCT compared with patients with slower ILC reconstitution dynamics.19 It remains to be investigated whether ILC, apart from maintaining and repairing epithelial barrier integrity, have additional modes of action to protect against GVHD.
Here, we report the presence of a regulatory subset of human, CD39+CD73+ ectoenzyme-expressing (ecto+) ILC3 with immunosuppressive properties. Ecto+ ILC3 converted eATP into adenosine, and in coculture experiments, ecto+ ILC3 suppressed T-cell proliferation. GVHD was associated with lower serum levels of adenosine, and in gut biopsies of patients with GVHD, proportions of ecto+ ILC3 were decreased when compared with noninflamed tissues. These data suggest that the predisposition of allo-HSCT recipients with delayed recovery of ILC to develop GVHD19 may be related to insufficient ecto+ ILC3 reconstitution and subsequent incapacity to neutralize the proinflammatory effects of DAMPs.
Materials and methods
Patients and healthy human materials
Study protocols were approved by the Medical Ethical Committee of Amsterdam UMC, location AMC. All participants signed informed consent. Tonsils were obtained from tonsillectomies. Healthy peripheral blood mononuclear cells were isolated from buffy coats from blood donations (Sanquin, Amsterdam, The Netherlands). Healthy bone marrow was obtained by sternum aspirate from patients undergoing cardiac surgery. Cord blood was obtained from umbilical cords after healthy deliveries at our center. Gut biopsies were obtained from the sigmoid colon of allogeneic HCT recipients (age, 52.3 ± 20.3 years) with diarrhea, suspected of stage 1 to 3 acute or late-onset acute GVHD of the gut. All patients included had biopsy-proven GVHD, and biopsies were taken at diagnosis, before initiation of corticosteroids. Noninflamed and inflamed colon tissue was obtained from patients undergoing colectomy for inflammatory bowel disease (IBD). Human fetal tissues were obtained from elective abortions at the Stichting Bloemenhove Clinic in Heemstede, The Netherlands, on receipt of informed consent.
Tissue collection and cell isolation
Tonsil tissue was cut in small pieces and mechanically disrupted, using the Stomacher 80 Biomaster. Cell suspensions were filtered through a 70-μm cell strainer, and mononuclear cells were isolated with Ficoll-Paque Plus medium (GE Healthcare). Before sorting tonsillar ILC, mononuclear cell samples were depleted of T cells and B cells by labeling with fluorescein isothiocyanate (FITC)–conjugated anti-CD3 and anti-CD19, followed by anti-FITC microbeads (Miltenyi) according to the manufacturer’s instructions. Intestinal colon tissues were digested for 1 hour at 37°C with collagenase D (60 U/mL) and DNase I (50 U/mL) in medium (phosphate-buffered saline; 500 μM Mg2+, 150 μM Ca2+, 1 mM pyruvate, pen/strep), and passed through a 70-μm cell strainer. Serum was collected by centrifugation of whole blood, and peripheral blood mononuclear cells were isolated by Lymphoprep (Stemcell Technologies) density gradient centrifugation. Total ILC were defined as lineage negative (CD1a, CD3, CD4, CD8, CD14, CD16, CD19, CD34, CD94, CD123, BDCA2, FcεRI, TCRαβ, TCRγδ), CD45+, CD127+, and CD161+. ILC1 were defined as CD117−, ILC2 as CRTH2+, and ILC3 as CD117+CRTH2−. Cells were sorted on a FACSAria (BD) to a purity higher than 99%.
Conjugated antibodies
FITC-conjugated anti-CD1a (HI149), anti-CD3 (OKT3), anti-CD14 (HCD14), anti-CD16 (3G8), anti-CD19 (HIB19), anti-CD94 (DX22), anti-CD123 (6H6), anti-FcεR1α (AER-37), anti-TCRαβ (IP26), anti-TCRγδ (B1; all from BioLegend); FITC-conjugated anti-CD34 (581; BD Biosciences), anti-BDCA2 (CD303, Miltenyi Biotec), anti-CRTH2 (CD294, BM16; BD Biosciences); phycoerythrin (PE)-indotricarbocyanine (Cy7)–conjugated anti-CD127, PE-Cy5.5–conjugated anti-CD117 (both from Beckman Coulter); Alexa Fluor 700 (AF700)–conjugated anti-CD3 (UCHT1), APC-Cy7–conjugated anti-CD45 (2D1), Brilliant Violet 421 (BV421)–conjugated anti-CD73 (AD2), BV510-conjugated anti-CD161 (HP-3G10), PE-Cy5.5–conjugated anti-CD4 (OKT4), PE-conjugate anti-CD5 (UCHT2; all from BioLegend); and PE-Texas Red-conjugated anti-CD39 (TU66; BD Biosciences). For phenotypical analyses by flow cytometry on BD LSR II Fortessa, the following antibodies were used: allophycocyanin (APC)-conjugated anti-CD25 (BC96; eBioscience), AF700-conjugated anti-CD69 (FN50; BioLegend); PE-conjugated anti-CD38 (HB7; BD Biosciences), APC-conjugated anti-CD26, PE-conjugated anti-CD103, APC-conjugated anti-CD25 (BC96; eBioscience), anti-CD45RO (UCHL1; BioLegend); PE-conjugated anti-CD45RA (IM1834U; Beckman Coulter), and anti-NKp44 (CD336, P44-8; Sony Biotechnology).
Cytokine stimulation and intracellular cytokine staining
Populations of ILC were cultured in a round-bottom 96-well plate for 1 or 3 days in Yssel’s medium, supplemented with 1% heat-inactivated human AB serum (Merck) and 1% penicillin and streptomycin (Roche) with IL-2 (10 U/mL) and in combination with IL-1β, IL-23 (50 ng/mL), or RA (1 μM; MilliporeSigma). For FACS analysis of cell surface markers, cells were stained with antibodies for 20 minutes at 4°C in phosphate-buffered saline. For intracellular cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate (10 ng/mL; MilliporeSigma) plus Ionomycin (500 nM; Merck) in the presence of GolgiPlug (BD) for 4 hours at 37°C. Thereafter, cells were fixed and permeabilized according to the manufacturer’s instructions (Invitrogen) and stained with following antibodies: AF700-conjugated anti–IL-17A (BL167), FITC-conjugated anti–IL-22 (22URT1), and PE-Cy7–conjugated anti-IFN-γ (all from BD Biosciences). Phenotypical analyses were performed by flow cytometry on LSR II Fortessa (BD Biosciences).
T-cell proliferation assay
T cells were isolated from tonsil, using PE-Cy5.5–conjugated anti-CD4 and PE-conjugate anti-CD5 (UCHT2; BioLegend). Autologous CD4+CD5+CD39− T-cell proliferation was assessed by CellTrace Violet dilution assay. Briefly, 20 000 cells were stained with Cell-Trace Violet Cell Proliferation Dye (C34557, Invitrogen; 20 minutes at 37°C), washed, and then cultured in RPMI 1640/10% fetal bovine serum at 37°C, in the presence of anti-CD3/anti-CD28 monoclonal antibody-coated beads (Dynabeads Human, T-activator CD3/CD28; Thermo Fisher Scientific). The next day, stimulated CD4+ T cells were cultured in the presence or absence of autologous ILC (1:1 ratio) and in the presence or absence of ATP (MilliporeSigma) in Yssel’s medium (in 96-well plates) with 5% fetal calf serum and penicillin-streptomycin. After 3 or 5 days, cells were harvested, stained with anti-CD4 and anti-CD39, and analyzed on LSR Fortessa (BD Biosciences).
Real-time quantitative polymerase chain reaction
Total RNA was extracted with a NucleoSpin RNA XS Kit (Macherey-Nagel) according to the manufacturer’s instructions. cDNA was synthesized with a high-capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Polymerase chain reactions were performed in BioRad iCycler (BioRad) with IQ SYBR Green Supermix (Bio-Rad), using the primers described in supplemental Table 1. Bio-Rad CFX Manager 3.1 software was used for quantification of expression. All samples were normalized to the expression of the housekeeping gene GAPDH, and results are presented in arbitrary units.
ATPase activity and HPLC tandem-MS
ATP (MilliporeSigma) and sodium polyoxotungstate 1 (POM-1; Tocris Bioscience) were dissolved in water. ATPase/GTPase Activity Assay Kit (MilliporeSigma) colormetric detection of phosphate with malachite green reagent was used according to the manufacturer’s instructions. Two hundred microliters supernatant from cells (20 000 cells seeded in 96-well plate) stimulated for 24 hours in the presence or absence of eATP was stored at −80°C and used for high-performance liquid chromatography (HPLC) tandem-mass spectrometry (MS) analysis of adenosine and inosine concentrations. HPLC tandem-MS analysis were performed as previously described.20 The analysis was performed with the following alterations: samples were deproteinized with a 30-kDa filter and analyzed using Waters Premier XE, a Waters Acquity Ultra Performance Liquid Chromatography (UPLC) pump, a Waters Acquity injector, and an analytical column (Waters HSS T3; 100 × 2.1 mm, 1.8 µM, injection volume: 10 µL).
Statistical analysis
Statistical significance was determined using GraphPad Prism software with a Student t test for paired samples, or analysis of variance test when groups of samples were analyzed. Values of P < .05 were considered to be significant.
Results
Ectoenzymes are expressed by ILC3 in the oral-gastrointestinal tract and in the bone marrow
First, we investigated whether human ILC express ectoenzymes. Flow cytometric analysis of CD45+CD127+CD161+ ILC subsets (Figure 1A) derived from tonsils, bone marrow, fetal intestine, and fetal mesenteric lymph nodes (mLNs) showed that ILC3 (CD117+CRTH2−) from tissues and bone marrow expressed several adenosine-generating enzymes, including the tri- and diphosphohydrolase (NTPDase) CD39, the 5′-ectoenzyme (5′-NT) CD73, and the NAD+ nucleosidase CD38 (Figure 1). CD117+CRTH2− ILC that contain ILC3 precursors from healthy peripheral blood and cord blood expressed very low levels of CD39, CD73, and CD38. This was also the case for ILC1 and ILC2 from different tissues (including tonsils, bone marrow, fetal intestine, and fetal mLN) and blood (supplemental Figure 1A-B). In contrast, subpopulations of ILC3 from bone marrow and tissues associated with the oral-gastrointestinal tract (tonsils, mLN, and intestines) did express ectoenzymes, with the highest frequencies observed in the fetal intestine (Figure 1B). There was a low variation in expression of CD39 and CD73 on ILC3 across individuals. In tonsil, for instance, CD39+CD73+ double-positive ILC3, which we dubbed ecto+ ILC3, represented 15,5% (standard deviation [SD], ±3.5; n = 25). CD39−CD73− double-negative (DN) ILC3 represented 44.0% (SD, ±5.9) of the total ILC3 population (Figure 1C). The variation in the presence of single positive CD39+CD73− ILC3 and CD39−CD73+ ILC3 was higher, and these subsets represented, on average, 22.7% (SD, ±9.2) and 17.4% (SD, ±8.1), respectively, of the total ILC3 population.
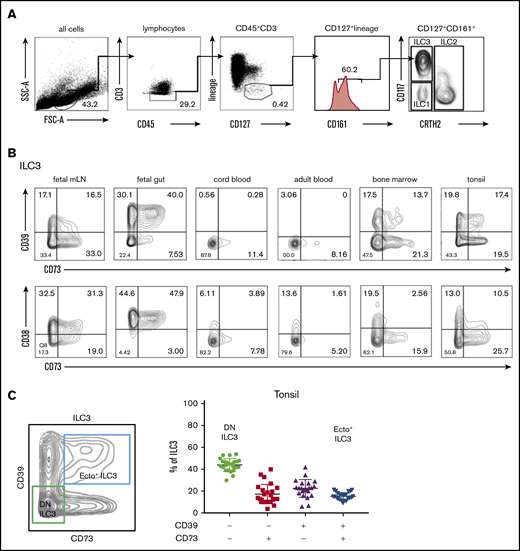
Flow cytometric analysis of ectoenzyme expression on ILC across different tissues. (A) Gating strategy for flow cytometric analysis of human ILC from healthy peripheral blood, with ILC defined as: Lin− (CD1a, CD3, CD4, CD8, CD14, CD16, CD19, CD34, CD94, CD123, BDCA2, FcεRI, TCRαβ, TCRγδ), CD45+ CD127+CD161+ cells. From this population, CRTH2+ cells define ILC2; CRTH2−CD117−NKp44− cells define ILC1, and CD117+ cells define ILC3. (B) CD39-CD73 and CD38-CD73 expression by ILC3 freshly isolated from fetal intestine and mLNs, cord blood, adult peripheral blood, healthy bone marrow, and tonsils. Contour plots are representative of 2 to 3 independent experiments. (C) Contour plot showing CD39−CD73− (DN) ILC3 and CD39+CD73+ (ecto+) ILC3 gating in tonsils, and the bar graph shows the frequency of CD39 and CD73 on total tonsillar ILC3 (n = 25); each symbol represents an individual donor, error bars show the standard error of the mean (SEM), and the horizontal lines represent the mean.
Flow cytometric analysis of ectoenzyme expression on ILC across different tissues. (A) Gating strategy for flow cytometric analysis of human ILC from healthy peripheral blood, with ILC defined as: Lin− (CD1a, CD3, CD4, CD8, CD14, CD16, CD19, CD34, CD94, CD123, BDCA2, FcεRI, TCRαβ, TCRγδ), CD45+ CD127+CD161+ cells. From this population, CRTH2+ cells define ILC2; CRTH2−CD117−NKp44− cells define ILC1, and CD117+ cells define ILC3. (B) CD39-CD73 and CD38-CD73 expression by ILC3 freshly isolated from fetal intestine and mLNs, cord blood, adult peripheral blood, healthy bone marrow, and tonsils. Contour plots are representative of 2 to 3 independent experiments. (C) Contour plot showing CD39−CD73− (DN) ILC3 and CD39+CD73+ (ecto+) ILC3 gating in tonsils, and the bar graph shows the frequency of CD39 and CD73 on total tonsillar ILC3 (n = 25); each symbol represents an individual donor, error bars show the standard error of the mean (SEM), and the horizontal lines represent the mean.
Ecto+ ILC3 express purinergic receptors and display a tissue-resident phenotype
We then further characterized phenotypical differences between tonsillar ecto+ ILC3 and DN ILC3. First, we analyzed expression of a selection of purinergic P1 and P2 receptors. As the gene expression of these receptors by ILC is unknown, we compared their expression on ILC3 with conventional CD4+CD39− T cells and CD4+CD25+CD39+ T cells that are enriched for Treg cells. The gene expression of P2X7R was not significantly different on ecto+ ILC3 and DN ILC3, but P2X7R was significantly higher in ecto+ ILC3 compared with CD39+ T cells (Figure 2A). P2Y11R expression was significantly higher in ecto+ ILC3 compared with DN ILC3 and CD4+ T cells, but comparable to CD39+ T cells (Figure 2A). The gene expression of the purinergic P1 receptor A2A (Ado2aR) was similar on both ILC3 subsets and significantly lower compared with CD4+ T cells and CD39+ T cells (Figure 2A).
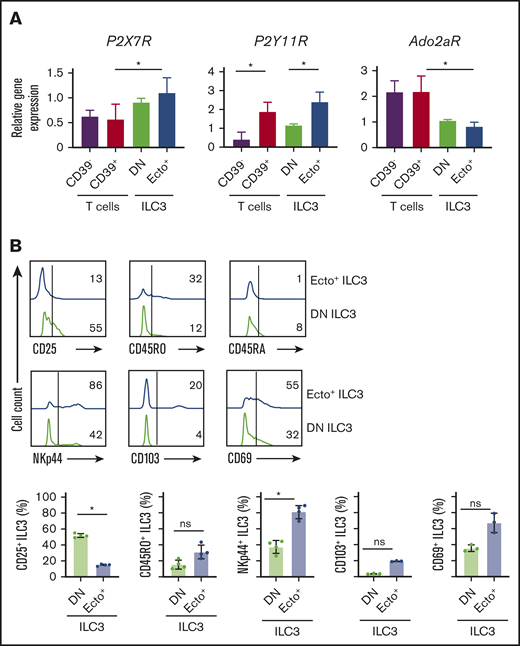
Phenotypical differences. (A) P2X7R, P2Y11R, and Ado2aR relative gene expression by tonsillar CD39−CD73− (DN) ILC3, CD39+CD73+ (ecto+) ILC3, CD4+CD39− T cells, and CD4+CD39+ T cells. Data are normalized to the expression of GAPDH. Data are presented as mean ± SEM. *P < .05. Data are summarized from or representative of 3 to 4 independent experiments. (B) Representative flow cytometric analysis of CD25, CD45RO, CD45RA, NKp44, CD103, and CD69 on ecto+ ILC3 (blue line) and DN ILC3 (green line). Horizontal line represents isotype control. Quantification is shown in the lower panel. Data are representative of as individual values of 3 to 4 independent experiments; *P < .05 by Student t test.
Phenotypical differences. (A) P2X7R, P2Y11R, and Ado2aR relative gene expression by tonsillar CD39−CD73− (DN) ILC3, CD39+CD73+ (ecto+) ILC3, CD4+CD39− T cells, and CD4+CD39+ T cells. Data are normalized to the expression of GAPDH. Data are presented as mean ± SEM. *P < .05. Data are summarized from or representative of 3 to 4 independent experiments. (B) Representative flow cytometric analysis of CD25, CD45RO, CD45RA, NKp44, CD103, and CD69 on ecto+ ILC3 (blue line) and DN ILC3 (green line). Horizontal line represents isotype control. Quantification is shown in the lower panel. Data are representative of as individual values of 3 to 4 independent experiments; *P < .05 by Student t test.
In addition, we analyzed the expression of markers associated with tissue residency and activation status on ecto+ ILC3 and DN ILC3 (Figure 2B). The ecto+ ILC3 population contained a significantly higher frequency of NKp44 and a trend toward higher CD45RO-expressing cells, whereas both subsets were mainly negative for CD45RA (Figure 2B) and HLA-DR (data not shown), and the DN ILC3 population contained significantly more CD25+ cells. Furthermore, ecto+ ILC3 had a trend toward higher expression of markers characteristic for tissue-resident lymphocytes, including CD103 and CD69 (Figure 2B), respectively. Of note, the expression of the ILC3 signature transcription factor RORC was similar in ecto+ ILC3 and DN ILC3 subsets (supplemental Figure 2A-B), confirming that these ecto+ ILC3 are bona fide ILC3. Together, these data suggest that ecto+ ILC3 have a different activation status compared with DN ILC3.
ILC3 ectoenzyme expression is regulated by IL-1β
In mucosal tissues, ILC3 activation is induced in response to the APC-derived mediators IL-1β, IL-23, and RA. To analyze the effect of these mediators on ectoenzyme expression of ILC3, we incubated ILC3 from tonsil for 1 or 3 days in the presence of IL-1β, IL-23, or RA, and analyzed expression of CD39 and CD73 by flow cytometry (Figure 3). In tonsils, the majority of ecto+ ILC3 coexpressed NKp44 (Figure 3). When compared with either freshly isolated tonsillar ILC3, medium-only or IL-2-only conditions, stimulation with IL-1β induced the coexpression of CD39 and CD73 both on NKp44− ILC3 (Figure 3C), as well as on NKp44+ ILC3 (Figure 3D), whereas RA induced the expression of CD73 on predominantly on NKp44+ ILC3 (supplemental Figure 3A). There was no significant difference in absolute numbers comparing the conditions with or without IL-1β stimuli (supplemental Figure 3B), suggesting that the phenotypical changes we observed are a result of changes in relative frequencies, and not actual expansion of specific subsets. To address which ILC3 subset responded to IL-1β, we sorted DN ILC3, ecto+ ILC3, CD39+ SP ILC3, and CD73+ ILC3 and observed that predominantly CD73+ SP ILC3 responded by upregulating CD39 expression and becoming ecto+ ILC3 (supplemental Figure 3C). The addition of IL-23 together with IL-1β or stimulation with eATP had no significant effect on the expression of CD39 and CD73 on ILC3 (data not shown).
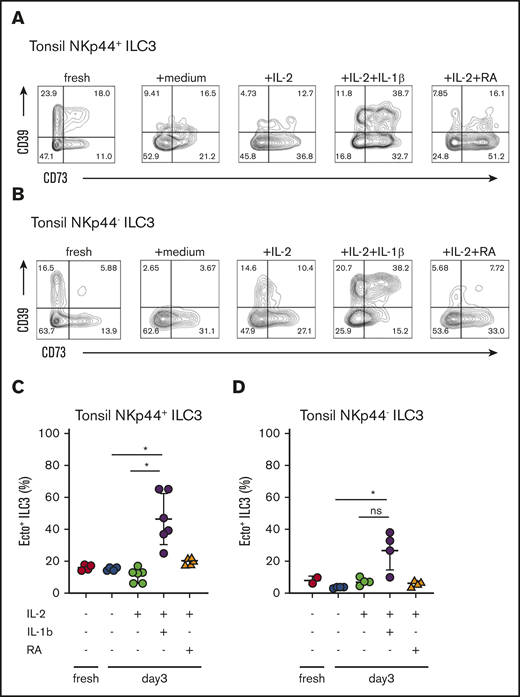
Cytokines that upregulate and/or maintain ectoenzymes on human ILC3. Cell surface expression of CD39 and CD73 on tonsillar NKp44+ ILC3 (A) and tonsillar NKp44− ILC3 (B) freshly isolated; after exposure to no cytokines (medium condition); IL-2 alone; IL-2 and IL-1β; IL-2 and RA for 3 days. Contour plots are representative of 3 independent experiments, including experimental duplicates. (C) Quantification of CD39 and CD73 coexpression on tonsillar NKp44+ ILC3 after 3 days of exposure with mediators. Data represent at least 4 independent experiments, including duplicates. Error bars show SEM, and horizontal bars show means. *P < .05 as determined by 1-way analysis of variance or Student t test.
Cytokines that upregulate and/or maintain ectoenzymes on human ILC3. Cell surface expression of CD39 and CD73 on tonsillar NKp44+ ILC3 (A) and tonsillar NKp44− ILC3 (B) freshly isolated; after exposure to no cytokines (medium condition); IL-2 alone; IL-2 and IL-1β; IL-2 and RA for 3 days. Contour plots are representative of 3 independent experiments, including experimental duplicates. (C) Quantification of CD39 and CD73 coexpression on tonsillar NKp44+ ILC3 after 3 days of exposure with mediators. Data represent at least 4 independent experiments, including duplicates. Error bars show SEM, and horizontal bars show means. *P < .05 as determined by 1-way analysis of variance or Student t test.
Ecto+ ILC3 have ATPase activity and hydrolyze eATP into adenosine
The proinflammatory effects of DAMPs such as eATP and NAD+ are neutralized by ectoenzyme-expressing immune cells that convert these DAMPs into adenosine, which has immunosuppressive properties. To evaluate the enzymatic activity of ecto+ ILC3, we measured ATPase activity and adenosine levels in ecto+ ILC3 supernatants after treatment with eATP. To block enzymatic activity (phosphohydrolysis of ATP), we used the pharmacologic CD39 inhibitor POM-1 Extracellular ATP (used at 50 µM) and POM-1 (used at 100 µM) were added to the culture at a concentration that did not affect ILC survival (measured by annexin V negative staining; Figure 4A). Culture of ecto+ ILC3 and DN ILC3 in the presence of eATP showed that ecto+ ILC3 and CD39+CD73− ILC3 have ATPase activity (6.6 ± 0.25 µMol/min per µL; n = 3) similar to CD39+ T cells (7.15 ± 1.20 µMol/min per µL), whereas DN ILC3 did not exhibit significant ATPase activity (Figure 4B). The ATPase activity of ecto+ ILC3, similar to that of CD39+ T cells, could partially be inhibited by POM-1 (4.6 ± 0.85 µMol/min per µL; Figure 4B). Next, we performed HPLC tandem-MS to measure adenosine in supernatants of ecto+ ILC3 cultured in the presence or absence of eATP. Ecto+ ILC3 were able to hydrolyze eATP into adenosine (0.266 ± 0.128 µMol/L; n = 4), and this capacity was significantly suppressed by treatment with POM-1. In the culture supernatants of DN ILC3 stimulated with eATP, adenosine levels were below detection threshold (Figure 4C). Interestingly, although CD39+ Treg cells had similar ATPase activity as ecto+ ILC3 (Figure 4B), Treg cells did not produce similar adenosine levels (below detection threshold) as ecto+ ILC3 (Figure 4C). This may be related to the lack of CD73 expression, and hence AMPase activity of CD39+ Treg cells, whereas ecto+ ILC3 express both ectoenzymes required for the hydrolysis of eATP into adenosine. Indeed, ectoenzyme SP ILC3 lacked either ATPase (CD39−CD73+ ILC3) or AMPase activity (CD39+ CD73− ILC3) and were not able to convert ATP into adenosine (data not shown).
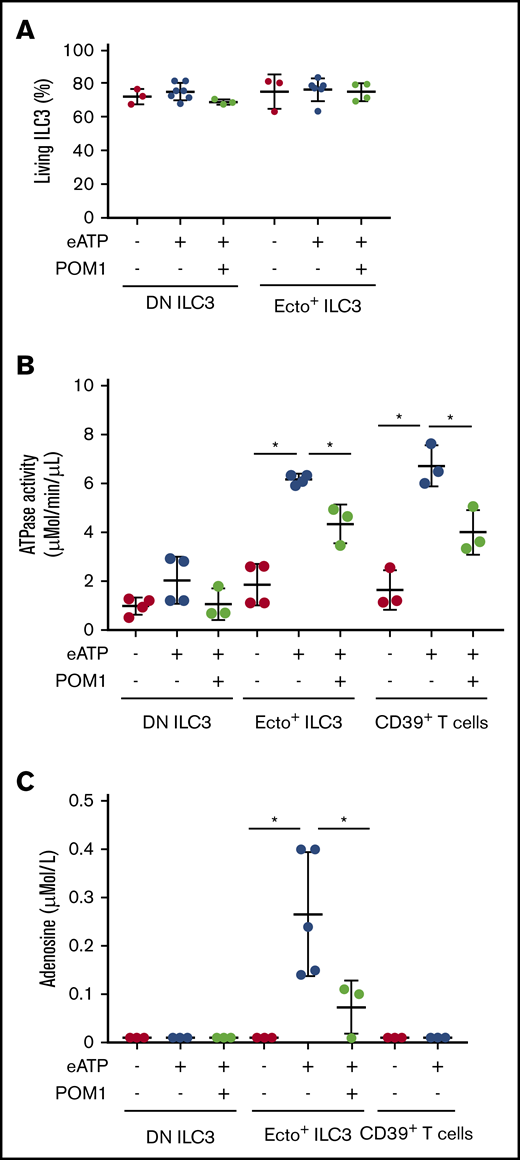
ATPase activity and adenosine measurements. (A) Flow cytometric analysis of annexin V staining (percentage of annexin V negative cells) showing cell survival before and after eATP (used at 50 µM) or sodium POM-1 (used at 100 µM) treatment. (B) ATPase activity (µMol/min per µL) after 30 minutes of exposure to eATP or eATP plus CD39 inhibitor POM-1. (C) HPLC tandem-MS adenosine measurement (µMol/L) before and after exposure to eATP or eATP plus POM-1. Data represent 3 to 5 independent experiments. Error bars show SEM, and horizontal bars show means. *P < .05 as determined by Student t test.
ATPase activity and adenosine measurements. (A) Flow cytometric analysis of annexin V staining (percentage of annexin V negative cells) showing cell survival before and after eATP (used at 50 µM) or sodium POM-1 (used at 100 µM) treatment. (B) ATPase activity (µMol/min per µL) after 30 minutes of exposure to eATP or eATP plus CD39 inhibitor POM-1. (C) HPLC tandem-MS adenosine measurement (µMol/L) before and after exposure to eATP or eATP plus POM-1. Data represent 3 to 5 independent experiments. Error bars show SEM, and horizontal bars show means. *P < .05 as determined by Student t test.
Ecto+ ILC3 suppress T-cell proliferation
To investigate whether ecto+ ILC3 have immunosuppressive capabilities, ILC3 were cocultured with T cells. We focused in these functional experiments on DN ILC3 and ecto+ ILC3, as ectoenzyme SP ILC3 did not have adenosine producing capacities. T cells were sorted as CD4+CD5+CD39− cells and activated with anti-CD3/CD28 beads. T-cell proliferation, as measured by the dilution of CellTrace Violet using flow cytometry, was significantly inhibited when cocultured with autologous ecto+ ILC3 preactivated with eATP (Figure 5A). T-cell proliferation was unchanged when cocultured with DN ILC3, either in the presence or absence of eATP or when cultured with eATP only (Figure 5A). Adenosine levels in supernatants of ecto+ ILC3 cocultured with T cells were below the level of detection (data not shown), suggesting that adenosine may be bound to adenosine receptor-expressing T cells to exert its suppressive effect. Alternatively, adenosine may also be deaminated to anti-inflammatory metabolite purine nucleoside inosine by CD26 in complex with the ADA receptor. Inosine measured by HPLC tandem-MS was detectable when ecto+ ILC3 were cocultured with T cells in the presence of eATP (Figure 5B). As expected, inosine was not detected when activated T cells were cocultured with DN ILC3 and eATP (data not shown), as adenosine cannot be formed in the absence of CD39 and CD73. ADA mRNA was expressed in ecto+ ILC3, but at lower levels compared with DN ILC3 or CD4+ T cells (Figure 5C). In addition, flow cytometric analysis of tonsillar ILC3 showed that CD26 is significantly lower expressed on ecto+ ILC3 compared with DN ILC3 (Figure 5D). Together, these data reveal that adenosine producing ecto+ ILC3 have the capacity to suppress proliferation of activated T cells. The reduced expression of both ADA and CD26 may endow ecto+ ILC3 with the ability to concentrate adenosine and use it for immune suppression.
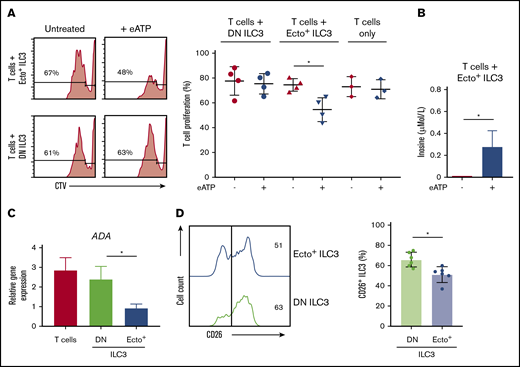
T-cell proliferation. (A) Representative flow cytometric analysis (left panel) and quantification (right panel) of CellTrace Violet stain showing T-cell proliferation by CD4+CD5+CD39- T cells alone or after coculture with DN ILC3 or ecto+ ILC3, in the presence or absence of eATP. (B) HPLC tandem-MS inosine measurement (µMol/L) of ecto+ ILC3 cocultured with CD4+CD5+CD39- T cells untreated or eATP-stimulated. (C) ADA gene expression by tonsillar DN ILC3, ecto+ ILC3 and CD4+ T cells. Data are presented as mean ± SEM. Data are summarized from 3-4 independent experiments. (D) CD26 expression on ecto+ ILC3 (blue line) and DN ILC3 (green line) from tonsils and quantified by comparing paired samples (n = 6, right panel). *P < .05 as determined by Student t test.
T-cell proliferation. (A) Representative flow cytometric analysis (left panel) and quantification (right panel) of CellTrace Violet stain showing T-cell proliferation by CD4+CD5+CD39- T cells alone or after coculture with DN ILC3 or ecto+ ILC3, in the presence or absence of eATP. (B) HPLC tandem-MS inosine measurement (µMol/L) of ecto+ ILC3 cocultured with CD4+CD5+CD39- T cells untreated or eATP-stimulated. (C) ADA gene expression by tonsillar DN ILC3, ecto+ ILC3 and CD4+ T cells. Data are presented as mean ± SEM. Data are summarized from 3-4 independent experiments. (D) CD26 expression on ecto+ ILC3 (blue line) and DN ILC3 (green line) from tonsils and quantified by comparing paired samples (n = 6, right panel). *P < .05 as determined by Student t test.
Extracellular ATP triggers IL-22 secretion by ILC3
An important characteristic of activated ILC3 is their ability to produce IL-22, which is a cytokine that plays a key role in epithelial tissue homeostasis and repair. To address the question of whether eATP hydrolysis by ecto+ ILC3 would affect their tissue regenerative capabilities, we examined the effect of eATP on IL-22 production by ILC3. Twenty-four hours of eATP treatment, followed by stimulation with phorbol 12-myristate 13-acetate plus ionomycin, significantly enhanced IL-22 production by both ecto+ ILC3 and DN ILC3 (Figure 6). This increase of IL-22 production by both subsets may hence be more indicative of the involvement of the ATP-sensing purinergic P2 receptors, for instance P2X7R or P2Y11R, which are expressed by both ILC3 subsets (Figure 2A), rather than a role for ectoenzymes. No effect of eATP on IL-17 production by ILC3 was observed (Figure 6). These data suggest that tissue damage leading to release of DAMPs, such as ATP, may boost ILC3 to produce more IL-22 for epithelial tissue repair.
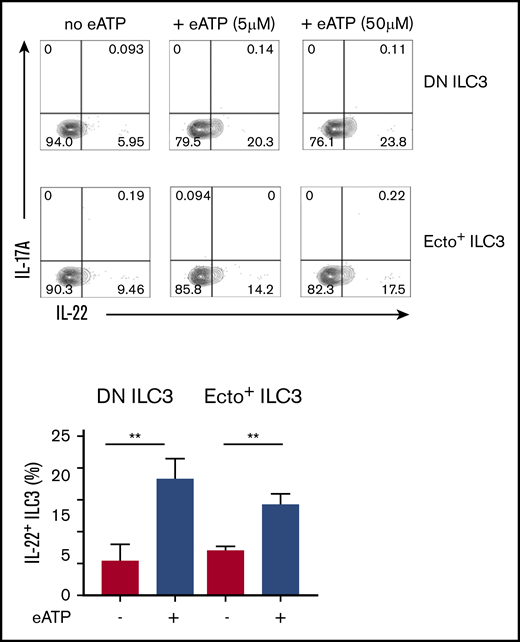
Effects of extracellular (e)ATP. Representative flow cytometric analysis (upper panel) and quantification (lower panel) of intracellular IL-22-producing DN ILC3 and ecto+ ILC3, on 3 days exposure to eATP. Data are presented as mean ± SEM; **P < .01 as determined by Student t test. Data are summarized from 3-4 independent experiments.
Effects of extracellular (e)ATP. Representative flow cytometric analysis (upper panel) and quantification (lower panel) of intracellular IL-22-producing DN ILC3 and ecto+ ILC3, on 3 days exposure to eATP. Data are presented as mean ± SEM; **P < .01 as determined by Student t test. Data are summarized from 3-4 independent experiments.
Ecto+ ILC3 are decreased in the gut of patients with GVHD
Tissue damage, release of DAMPs, alloreactive T-cell activation, and lack of negative feedback mechanisms on immune activation are key features in GVHD pathogenesis. In line with this, we observed that the levels of adenosine and its metabolite inosine, as measured by HPLC tandem-MS, in serum of patients with GVHD was significantly reduced compared with those of healthy subjects (Figure 7A-B). To address whether loss of ecto+ ILC3 may contribute to reduced adenosine/inosine levels, we analyzed the presence of ecto+ ILC3 in sigmoid biopsies from allo-HSCT patients at diagnosis of GVHD (supplemental Figure 4A). As a control, noninflamed and inflamed colon tissue was obtained from patients undergoing colectomy for IBD. Although the total frequency of NKp44− ILC3 and NKp44+ ILC3 in intestinal acute GVHD and noninflamed colon tissue of patients with IBD was similar (supplemental Figure 4B), a significant decrease in the proportion of ecto+ ILC3 (significantly decreased among the NKp44+ ILC3; supplemental Figure 4C) was found in GVHD-affected gut tissues compared with in noninflamed colon tissue (Figure 7C). Similarly, inflamed colon tissue of patients with IBD also showed a significant decrease in ecto+ ILC3, as observed in GVHD-affected tissues. Collectively, our data suggest that in GVHD, the capacity to neutralize eATP is limited by the reduced proportions of ecto+ ILC3 in tissues, limiting the capacity to prevent and repair inflammation related tissue damage.
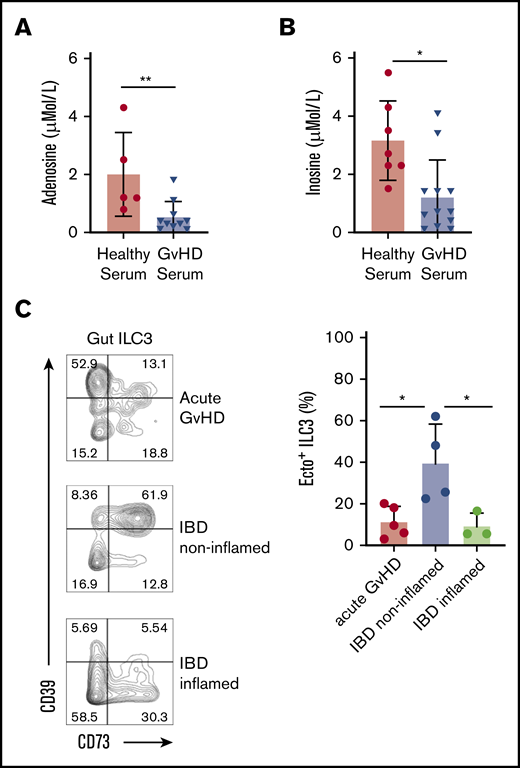
Ectoenzyme-expressing ILC in GVHD-affected gut tissue. HPLC tandem-MS measurements of serum adenosine (µMo/L) (A) and inosine (µMol/L) (B) from healthy subjects (n = 5) and patients affected by GVHD (n = 10). (C) Representative flow cytometric analysis (left panel) of CD39 and CD73 expression on gut ILC3 from patients with GVHD, inflamed and noninflamed gut tissues from patients with Crohn’s disease. Histograms shows quantification (right panel) of ecto+ ILC3 from patients with GVHD, inflamed and noninflamed control gut tissue tissues from patients with Crohn’s disease. Data represent 3 to 5 independent experiments. Error bars show SEM, and horizontal bars show means. *P < .05 as determined by 1-way analysis of variance or Student t test.
Ectoenzyme-expressing ILC in GVHD-affected gut tissue. HPLC tandem-MS measurements of serum adenosine (µMo/L) (A) and inosine (µMol/L) (B) from healthy subjects (n = 5) and patients affected by GVHD (n = 10). (C) Representative flow cytometric analysis (left panel) of CD39 and CD73 expression on gut ILC3 from patients with GVHD, inflamed and noninflamed gut tissues from patients with Crohn’s disease. Histograms shows quantification (right panel) of ecto+ ILC3 from patients with GVHD, inflamed and noninflamed control gut tissue tissues from patients with Crohn’s disease. Data represent 3 to 5 independent experiments. Error bars show SEM, and horizontal bars show means. *P < .05 as determined by 1-way analysis of variance or Student t test.
Discussion
GVHD develops when allogeneic immune activation outbalances tissue-restoring activities of the allogeneic and autologous immune systems. We have demonstrated that patients with rapid reconstitution of peripheral blood ILC numbers after acute myeloid leukemia induction chemotherapy, before allo-HSCT, had significantly less mucositis and were less prone to develop GVHD after allo-HSCT.19 However, it remains to be established how ILC mediate these tissue-protective effects. It has been demonstrated in mouse models that IL-22, produced by ILC, protected intestinal stem cells from apoptosis and thereby helped to maintain epithelial integrity in the gastrointestinal tract. Deficiency of IL-22 was associated with worsened GVHD, which could be reversed by IL-22 therapy.17 The question was raised, however, whether the protective effects of ILC solely depend on IL-22 production. Here, we demonstrated that CD39+CD73+ (ecto+) ILC3 represent a novel subset of innate lymphoid cells. Although CD39 expression was previously reported on ILC3,21 our data demonstrate that the co-expression of CD39 and CD73 identifies ILC3 that are functionally distinct compared with previously reported ILC3 subsets. Ectoenzyme-expressing T cells have been demonstrated to play an important role in limiting inflammation by hydrolyzing proinflammatory eATP into the potent anti-inflammatory mediator adenosine.3 In line with this, we found that ecto+ ILC3 were able to metabolize eATP into adenosine and suppress the proliferation of activated T cells, presumably through subsequent binding to the adenosine receptor. eATP-activated ILC3 produced increased levels of IL-22. Patients with acute GVHD of the intestine had significantly reduced proportions of ecto+ ILC3 in affected tissues compared to noninflamed tissue of patients with IBD, a similar pattern was observed in inflamed gut tissue from patients with IBD. These data offer a novel pathway, via which ILC may protect against the development of GVHD.
Expression of ectoenzymes can be regulated by proinflammatory cytokines, oxidative stress, and hypoxia, as has been demonstrated for T cells.22 In our hands, the proinflammatory cytokine IL-1β (abundant in inflamed gut), together with IL-2 increased the coexpression of the ectoenzymes CD39 and CD73 on ILC3. Low-dose IL-2 treatment has been reported to ameliorate GVHD, which could not solely be attributed to expansion or enhanced function of regulatory T cells,23 leaving room for other cells such as ecto+ ILC3 to contribute to the improvement of GVHD in these patients.
The effects of eATP on T cells are complex and depend on eATP concentration and on the variable expression of ATP-sensing receptors, including ectoenzymes and P2 receptors.24,25 For instance, P2X receptors are usually activated by lower eATP concentrations than P2Y receptors.26 It remains elusive, whether eATP differentially affects activation of ATP-sensing receptors resulting in altered ILC survival, activation, or function. We observed that stimulation of ILC3 with eATP, regardless of ectoenzyme expression, increased their IL-22-producing capacity. It is likely to assume that this is mediated by activation of P2 receptors, although it should be further investigated whether it is P2XR or P2YR activation.
Our findings are in line with the observation that deletion of the gene encoding for CD39, ENTPD1, exacerbates dextran-sulfate sodium-induced colitis in mice, and human ENTPD1 polymorphisms are associated with IBD susceptibility.27 Most recently, it was demonstrated that the ATP/CD39/adenosine pathway modulates ILC3 functions during tissue injury in mice.28 In patients with active ulcerative colitis, alteration in mucosal expression of genes involved in the purine metabolism was reported to correlate with upregulation of ILC3-IL-22 pathway signature genes. Researchers further showed in an experimental colitis model that pharmacological inhibition of the function of NTPDases (targeting NTPDase1/CD39 and NTPDase3) by POM-1 led to exacerbation of the colitis and reduced the number of IL-22-producing ILC3.28 Our data show that human ecto+ ILC are also receptive to the inhibitory effect of POM-1. Ex vivo treatment of human ecto+ ILC3 with POM-1 inhibited the ATPase activity of ecto+ ILC3, similar to that of human CD39+ T cells, suggesting a similar mechanism. Given the notion that CD39+ SP ILC3 with ATPase activity may also be involved in generating adenosine in concert with neighboring CD73+ cells; for instance, intestinal epithelial cells or CD73+ immune cells, it remains elusive what is the effect of ecto+ ILC3 in vivo. Further research in this direction should reveal whether an association exist between the severity of inflammatory diseases and the presence of ecto+ ILC3. It is evident, though, that ecto+ ILC3 can act independent of neighboring cells, and hence are able to promptly respond to environmental cues during which eATP levels increase.
On the basis of our observation that in response to eATP, ecto+ ILC3 produce immunosuppressive adenosine and IL-22, we propose that in allo-HSCT recipients, ILC3 may contribute to both the dampening of allogenic immune responses and to the restoration of tissue damage. The reduced frequency of ecto+ ILC3 in the gut of patients with GVHD, as compared with noninflamed gut, may contribute to the uncontrolled autologous T-cell responses, causing more damage. In line with this, we also detected equally low frequencies of ecto+ ILC3 in the gut of patients with active Crohn’s disease, in which tissue damage and deregulated immune responses are symptomatic for the disease as well. Moreover, we observed that serum levels of adenosine were significantly reduced in patients with GVHD, which coincides with low tissue numbers of ecto+ ILC3. Although we cannot exclude that increased uptake and degradation of adenosine is responsible for low adenosine serum levels, this may be unlikely, given the finding that inosine, the metabolite produced during the degradation of adenosine was also significantly reduced in the serum of patients with GVHD. It is tempting to conclude that reduced adenosine and inosine levels may reflect numerical deficits in ecto+ ILC3 in the context of GVHD. Either way, it is clear that the immune system of patients with GVHD has a reduced capacity to moderate inflammation related tissue damage.
In summary, we identified ecto+ ILC3 as adenosine-producing cells, with T-cell-suppressive capacities. The reduced frequency of gut ecto+ ILC3 in patients with GVHD indicate a reduced ability to dampen inflammation-related tissue damage, which may be a key factor in GVHD pathophysiology.
Acknowledgments
The authors thank the surgeons at the Onze Lieve Vrouwe Gasthuis (Amsterdam, The Netherlands) for providing the tonsillar tissue; the staff of the Bloemenhove Clinic (Heemstede, The Netherlands) for providing fetal tissues; and W. Kulik (Laboratory of Genetic Metabolic Diseases, Amsterdam UMC, location AMC) for measuring adenosine and inosine levels (by HPLC-MS/MS) in supernatant samples.
This research is financially supported by a Vidi grant (NWO ZonMW 91715362) and a Laboratory of Structural Biology Research Fellowship (1438F) (M.D.H.).
Authorship
Contribution: M.D.H. managed the recruitment of allo-HSCT recipients and patients with GVHD, contributed critical input, and wrote the paper; N.J.E.H. performed experiments; Y.F.v.L. managed the recruitment of patients with GVHD and contributed critical input; H.S. contributed critical input; L.K. helped with gut tissue processing; W.A.B. and C.J.B. managed the recruitment of patients with IBD; B.B. contributed critical input and wrote the paper; and M.M.S designed the study, performed experiments, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mette D. Hazenberg, Department of Hematology, Amsterdam Infection & Immunity Institute, Cancer Center Amsterdam, Meibergdreef 9 1105 AZ, Amsterdam, The Netherlands; e-mail: m.d.hazenberg@amsterdamumc.nl.

