

TO THE EDITOR:
The article by McMahon et al described 2 patterns of responses to gilteritinib treatment in acute myeloid leukemia (AML) patients with FLT3-internal tandem duplication (ITD) mutation: responses with and without hematopoietic differentiation.1 In this study, gilteritinib treatment resulted in myeloid differentiation in almost one-half of AML patients (10/21, 47.6%) with granulocytic hyperplasia without dysplastic changes. Interestingly, the proportion of erythroid lineage was unaffected or hypoplastic.
In contrast to those findings in the article, here we describe a unique differentiation pattern of myeloblasts into erythroid lineage induced by gilteritinib highlighting biological impacts of pharmacological blockade in FLT3 signaling.
A 63-year-old female with no significant medical history presented with fever, shortness of breath, and generalized weakness. Physical examination at presentation was unremarkable. A complete blood count showed a white blood cell count of 137.6 × 109/L with 38% blasts and 42% monocytes on differential, hemoglobin 8.0 g/dL, and platelet 72 × 109/L. A peripheral blood smear showed numerous monocytes with blasts (Figure 1A). A bone marrow biopsy revealed 95% hypercellular marrow and 86% myeloblasts with monocytic differentiation (Figure 1B). NPM immunostain of the biopsy demonstrated both nuclear and cytoplasmic NPM immunopositivity, indicating the presence of an NPM1 mutation (Figure 1C). Flow cytometry showed bright CD45; bright cytoplasmic myeloperoxidase; dim CD117, CD13, CD33; and dim CD4, HLA-DR, CD38. A small subset expressed CD34 (13%) and CD7 (12%). Cytogenetics showed a normal karyotype, and a molecular study demonstrated NPM1 and FLT3-ITD mutations. The mutated-to-unmutated peak height ratio for FLT3-ITD (FLT3-ITD allelic ratio) was 0.8. Unfortunately, midostaurin was not available to the patient during hospitalization for induction chemotherapy. Hence, the patient underwent induction chemotherapy with high-dose cytarabine and mitoxantrone. A bone marrow biopsy at day 14 postinduction showed no evidence of leukemia. The patient initially refused allogeneic hematopoietic stem cell transplantation (HCT) and elected consolidation chemotherapy alone with 4 cycles of high-dose cytarabine and midostaurin. At the end of consolidation therapy, her complete blood count showed white blood cells 3.12 × 109/L with hemoglobin 10.1 and platelet 56 × 109/L (Figure 1D); a bone marrow biopsy revealed hypercellular marrow (80%) and 47% pleiomorphic blasts in marrow aspirates (Figure 1E). Flow cytometry identified 30% of aberrant myeloblasts expressing CD117, CD13, CD33, and CD7 with a subset expressing CD34. Myeloid:erythroid ratio (ME ratio) was 0.4:1, with erythroid precursors 35%. Immunostain for NPM1 showed an abnormal pattern of nuclear and cytoplasmic staining, indicating presence of the mutation (Figure 1F). A variable allelic frequency of FLT3-ITD from peripheral blood at relapse was 21.5%, equivalent to allelic ratio 0.28, and the length of ITD insertion was 120 bp [FLT3 c.1837+12_1837+13ins(120)]. She was started on gilteritinib 120 mg daily and she tolerated the treatment well. A bone marrow biopsy after a 2-month gilteritinib treatment demonstrated a remarkable improvement; a peripheral blood smear failed to detect circulating blasts (Figure 1G) and the cellularity of bone marrow decreased to 30% with 1% blasts in the aspirates and abundance of dysplastic erythroid precursors (Figure 1H). Flow cytometry showed 2% of myeloblasts with aberrant CD7 expression. NPM immunostain of the biopsy again demonstrated the mutant staining pattern with nuclear and cytoplasmic reactivity in almost all of the marrow cells (Figure 1I), the majority of which were normoblasts with marked dysplasia in the aspirate smear. Interestingly, the distribution of erythroid precursors identified by E-cadherin2 and CD713 in the bone marrow before and after initiation of gilteritinib treatment were within NPM-stained areas, indicating that erythroid precursors were differentiated from mutant myeloblasts (Figure 1J-M). Furthermore, FLT3-ITD allelic ratio increased to 2.8 and the karyotype remained normal after initiation of gilteritinib. Notably, the ME ratio strikingly decreased to 0.2:1, with 80% of normoblasts indicating skewed differentiation of myeloblasts into normoblasts following gilteritinib treatment (Figure 2). The patient underwent matched unrelated peripheral blood HCT under a reduced intensity conditioning with fludarabine, melphalan, and total body irradiation following a 3-month course of gilteritinib treatment. A bone marrow biopsy 1 month after HCT revealed no morphologic or flow cytometric evidence of residual AML. Cellularity was 50% with trilineage maturation, and ME ratio was 1:1. Notably, NPM immunostain did not show aberrant cytoplasmic staining. A molecular study revealed no FLT3-ITD and tyrosin kinase domain mutations. The patient has currently been in complete remission at 80 days after HCT.
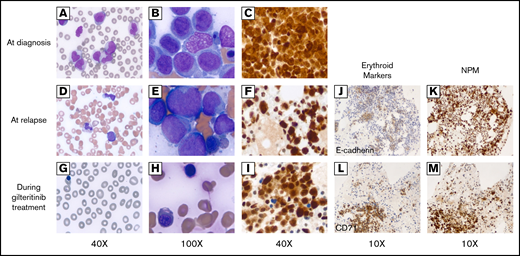
Peripheral blood smear and bone marrow biopsy before and after gilteritinib treatment. (A-C) Before gilteritinib treatment. (D-F) At relapse. (G-I) Two months after gilteritinib treatment. (A) Multiple monocytes and myeloblasts identified in peripheral blood smear (×40). (B) Clusters comprising blasts, monocytoid cells, and dysplastic normoblasts in bone marrow smear (×100). (C) NPM immunostain demonstrating both nuclear and cytoplasmic stains (×40). (D) A few monocytes and blasts in peripheral blood (×40). (E) Multiple monocytoid cells and blasts in clusters in bone marrow smear (×100). (F) Both nuclear and cytoplasmic stains by NPM immunostaining (×40). (G) Leukopenia without circulating blasts, polychromasia, and nucleated red blood cells in peripheral blood smear (×40). (H) Numerous erythroid precursors, dysplastic normoblasts identified in bone marrow smear (×100). (I) NPM immunostain redemonstrating abnormal staining pattern in both cytoplasms and nuclei (×40). (J) Erythroid precursors indicated by E-cadherin (×10) were within the distribution of NPM immunostain (×10) (K) at relapse. (L) Erythroid precursors indicated by CD71 (×10) that markedly increased after gilteritinib treatment were (M) a large proportion of NPM1-mutated cells in bone marrow.
Peripheral blood smear and bone marrow biopsy before and after gilteritinib treatment. (A-C) Before gilteritinib treatment. (D-F) At relapse. (G-I) Two months after gilteritinib treatment. (A) Multiple monocytes and myeloblasts identified in peripheral blood smear (×40). (B) Clusters comprising blasts, monocytoid cells, and dysplastic normoblasts in bone marrow smear (×100). (C) NPM immunostain demonstrating both nuclear and cytoplasmic stains (×40). (D) A few monocytes and blasts in peripheral blood (×40). (E) Multiple monocytoid cells and blasts in clusters in bone marrow smear (×100). (F) Both nuclear and cytoplasmic stains by NPM immunostaining (×40). (G) Leukopenia without circulating blasts, polychromasia, and nucleated red blood cells in peripheral blood smear (×40). (H) Numerous erythroid precursors, dysplastic normoblasts identified in bone marrow smear (×100). (I) NPM immunostain redemonstrating abnormal staining pattern in both cytoplasms and nuclei (×40). (J) Erythroid precursors indicated by E-cadherin (×10) were within the distribution of NPM immunostain (×10) (K) at relapse. (L) Erythroid precursors indicated by CD71 (×10) that markedly increased after gilteritinib treatment were (M) a large proportion of NPM1-mutated cells in bone marrow.
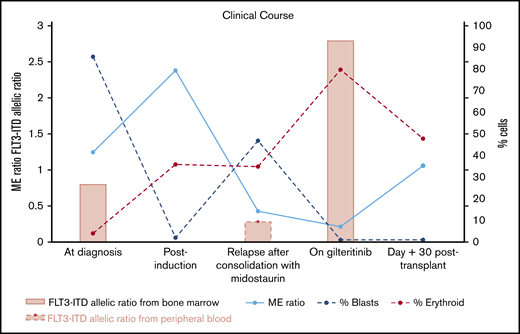
Summary of clinical courses with FLT3-ITD allelic ratio, ME ratio, and % blasts of bone marrow. ME ratio strikingly decreased by numerous erythroid precursors identified after initiation of gilteritinib treatment, whereas blasts significantly decreased. FLT3-ITD allelic ratio increased following gilteritinib treatment. Both FLT3-ITD and blasts became undetectable at day 30 after HCT.
Summary of clinical courses with FLT3-ITD allelic ratio, ME ratio, and % blasts of bone marrow. ME ratio strikingly decreased by numerous erythroid precursors identified after initiation of gilteritinib treatment, whereas blasts significantly decreased. FLT3-ITD allelic ratio increased following gilteritinib treatment. Both FLT3-ITD and blasts became undetectable at day 30 after HCT.
FLT3-ITD is a common molecular aberration identified in AML4 and is associated with arrest of myeloid differentiation via suppression of C/EPBα expression.5,6 The negative effect of FLT3-ITD on myeloid differentiation is further supported by the fact that FLT3 inhibitors, including quizartinib7 and sorafenib8 as well as gilteritinib,1 induced in vivo myeloid maturation in patients with FLT3-ITD mutation. In fact, no prior studies demonstrated that pharmacological blockade of FLT3 signaling induced skewed erythroid differentiation from the blasts. In our case, a striking decrease in ME ratio by increased normoblasts with NPM1 mutation following gilteritinib treatment and persistence of FLT3-ITD indicates that erythroid differentiation was originated from leukemic myeloblasts. Interestingly, our patient’s FLT3-ITD allelic ratio increased, whereas the proportion of blasts was markedly decreased following gilteritinib treatment. This is consistent with the study by McMahon et al, in which FLT3 mutation frequencies increased in 2 patients with the differentiation responses. The prior midostaurin treatment in our case did not induce myeloid nor erythroid differentiation despite midostaurin treatment targeting FLT3-ITD.9 This might be explained by myelosuppression resulted from potent blockade of KIT signaling by midostaurin but not with gilteritinib.
Why was the erythroid differentiation prominently observed in our case following FLT3 inhibition? What are the biological underpinnings? Injections with human FLT3 ligand to mice resulted in decreased erythropoiesis with loss of erythroid island macrophages in murine bone marrow.10 In a transgenic mouse model, BM erythroid progenitor numbers were 5.4-fold decreased along with a significant reduction in hematocrit in mice expressing the human FLT3 ligand (FLT3L-Tg mice) compared with wild-type mice.11 Moreover, the expression of Mpl and Klf1 (megakaryocyte/erythroid lineage genes) was increased with decreased expression of Mpo, Csf3r, and Cebpa (myeloid-specific genes) in murine hematopoietic stem cells (Lineage−SCA1+CD117+) of FLT3L-Tg mice, suggesting that overactivation of FLT3 signaling by increased access to FLT3L skewed hematopoiesis toward myelopoiesis at the expense of erythropoiesis and thrombopoiesis. Furthermore, mean serum FLT3L concentrations in patients with Fanconi or aplastic anemia were much higher than in healthy individuals.12 Taken together, it is plausible that selective blockade of FLT3 signaling by gilteritinib may induce rebound of erythroid differentiation previously suppressed by excessive FLT3 signaling by FLT3-ITD before gilteritinib treatment. The mechanistic underpinnings of erythroid differentiation induced by selective FLT3 signaling blockade need to be further elucidated. In AML patients with FLT3-ITD, higher allelic burden of FLT3-ITD and failure to achieve complete molecular remission before transplantation were associated with poorer clinical outcomes.13 Despite the persistence of FLT3-ITD following gilteritinib treatment without achieving complete remission before transplantation, the patient has remained in complete molecular remission.
As in our case, novel targeted therapeutic agents (eg, FLT3 inhibitors, IDH1/2 inhibitors14-16 ) have been reported to induce maturation from leukemic blasts. Therefore, this phenomenon may complicate the definition of complete remission and its clinical significance. The effect of differentiation induced by gilteritinib and other targeted agents on clinical outcomes warrants further investigations.
Acknowledgments:
This work was supported by the Rush University Medical Center and Rush University Cancer Center. H.D.Y. is supported by the Division of Hematology, Oncology and Cell Therapy at Rush University Medical Center. C.U. is supported by the Coleman Foundation at Rush University Medical Center.
Contribution: H.D.Y. and C.U. conceived the idea, generated and analyzed the data, conducted literature search, and wrote and edited the manuscript; I.M. provided figures and data of pathology, conducted literature search, and edited the manuscript; S.N., M.L., M.J.H., D.A.K., and A.V. conducted the literature search and edited the manuscript; and all authors approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hyun Don Yun, Division of Hematology, Oncology and Cell Therapy, Department of Medicine, Rush University, 1725 W Harrison, Suite 834, Chicago, IL 60612; e-mail: hyun_don_yun@rush.edu.

