

Key Points
FVL homozygous mice have increased thrombus size and reduced pulmonary embolism burden compared with WT mice.
FVL carriers develop more stable thrombi, which become clinical deep vein thrombosis more often than noncarriers, explaining the FVL paradox.

Abstract
Humans carrying the factor V Leiden (FVL) variant have a fivefold increased risk for venous thrombosis. However, incidence of deep vein thrombosis (DVT) is proportionally greater than that of pulmonary embolism (PE) in these individuals. This is known as the FVL paradox. We hypothesized that the rate of initial DVT development is similar in FVL and noncarriers, but thrombi in FVL carriers are more stable and develop into a clinically significant DVT more often than in noncarriers. To test this, we induced thrombi in the femoral vein of wild-type (WT), heterozygous (F5L/+), and FVL homozygous (F5L/L) mice. Using intravital microscopy, thrombus size and embolization were visualized and emboli in the lungs were quantified. Compared with WT, femoral vein thrombi in F5L/+ and F5L/L mice were larger and embolized less. Total and large embolic events, the percentage of thrombus that embolized, and PE burden were significantly decreased in F5L/L mice. This suggests that in noncarriers (reflected by WT), a minor injury initially resulting in a small DVT tends to remain small and asymptomatic because of the embolization of the otherwise growing thrombus. Alternatively, the same insult in people with FVL (reflected by F5L/L) leads to thrombus growth as a result of less embolization, and thus symptomatic DVT development.
Introduction
Activated factor V (FVa) together with the serine protease factor Xa (FXa) form the prothrombinase complex that converts prothrombin to active thrombin. FVa is subsequently inactivated by the natural anticoagulant activated protein C, which cleaves FVa at arginine(R)-506, R306, and R679 in the heavy chain1 . Cleavage at R506 is required for efficient cleavage at the 2 other sites.1 The substitution of glutamine (Q) for R506 in FV is a common mutation in humans known as factor V Leiden (FVL). The R506Q mutation abolishes a cleavage site for activated protein C, thereby reducing the rate of FVa inactivation and consequently allowing for prolonged prothrombinase activity.1
The clinical manifestation of the FVL variant varies. Whereas most individuals with FVL may never develop thrombosis,2 those with FVL heterozygosity are at increased risk for venous thrombosis, typically experiencing their first episode during adulthood,3 whereas those homozygous for FVL tend to present with their first episode at a younger age.4 The relative risk for venous thrombosis is increased three- to eightfold in those heterozygous for FVL.5,6,7 Although most studies report an increased venous thromboembolism risk in those homozygous for FVL, there is more discrepancy within the relative risk estimates (nine- to 80-fold)5,6,7
FVL heterozygosity is an inherited autosomal dominant condition that is identified in 20% to 50% of patients with venous thromboembolic diseases8,9 and 5% to 8% of the population in Canada.10 There is a higher incidence of DVT than PE in those with FVL, and a low prevalence of FVL among people who suffer a fatal PE.8,9 This has been referred to as the FVL paradox. Although several mechanisms and hypotheses have been studied, none have explained the biological basis for the FVL paradox.11,12,13
A study performed by van Langevelde et al evaluated whether common venous thrombosis risk factors have differential effect on DVT and PE.14 This study found that along with FVL, reproduction-related risk factors, such as pregnancy and use of oral contraceptives, as well as obesity, also showed a higher risk for DVT than PE. Meanwhile, pulmonary conditions, such as chronic obstruction pulmonary disease, pneumonia, and sickle cell disease, are a higher risk factor for PE, but have little or no effect on DVT. This suggests a biology that underlies whether the presentation of venous thromboembolism will be DVT or PE, thus understanding the FVL paradox may have broader implications.
On the basis of this previous work, we wondered whether the incidence of subclinical thrombus formation is similar among FVL carriers and noncarriers, but FVL carriers develop a symptomatic DVT as a result of enhanced thrombus stability. In contrast, the thrombus remains subclinical in noncarriers because of microembolization. Therefore, to explain the FVL paradox, we assessed DVT stability after a small injury in F5L/+ and F5L/L compared with wild-type (WT) mice.
Methods
FVL mice, gifted from the Ginsburg laboratory, were generated carrying the homologous mutation (R504Q) inserted into the endogenous murine F5 gene. Adult heterozygous (F5L/+) and homozygous (F5L/L) mice are viable and fertile and exhibit normal survival.15 Experiments were carried out using our in vivo mouse model of thrombus stability, as previously described with slight modifications.16 Previously, a 1 × 2 mm 1.8% ferric chloride (FeCl3)–soaked filter paper placed on the medial side of the femoral vein for 5 minutes was used to initiate nonocclusive thrombi in WT mice for 2 hours. This concentration and time of application of FeCl3 occluded the femoral vein of F5L/L mice before the 2-hour experiment window. Thus, we shortened the FeCl3-soaked filter paper application time to 3 minutes to initiate smaller, reproducible, and nonocclusive thrombi in both F5L/+ and F5L/L mice. Intravital videomicroscopy recorded embolic events leaving the thrombus and the thrombus sizes for 2 hours. Lungs were harvested, sectioned, and stained for presence of PE. All other labeling, quantification, and analysis has been previously described.16 Prothrombin fragment 1 + 2 (F1 + 2) levels were measured by enzyme-linked immunosorbent assay (Cloud Corp., Houston, TX), following the manufacturer’s instructions.
Results and discussion
Thrombi initially induced in the femoral vein of WT, F5L/+, or F5L/L mice were not significantly different from each other in size (data not shown). After 2 hours, the F5L/+ and F5L/L mice exhibited a significantly greater change in thrombus size from baseline compared with WT mice (Figure 1A). Emboli breaking off the proximal tip of the thrombus were recorded and manually counted. Both the total number and number of large embolic events were significantly more in WT mice compared with in F5L/+ and F5L/L mice (Figure 1B-C). The percentage of thrombus that embolized in the 2-hour observation period, a measure of DVT turnover, was significantly reduced in F5L/+ and F5L/L mice compared with WT mice (Figure 1D). Taken together, these data suggest that the small thrombus induced in FVL heterozygous or homozygous mice stabilized over time, with reduced embolization. This enabled greater thrombus growth, whereas the small thrombus in WT mice continuously embolized without increasing the net thrombus size.
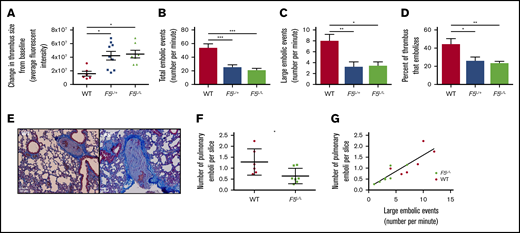
In vivo mouse model of thrombus stability in WT, F5L/+,and F5L/Lmice. The change in thrombus size from baseline (A), number of total (B) and large (C) embolic events, and percentage of thrombus that embolized (D). (E) Representative emboli images found in the pulmonary arteries of WT and F5L/L mice. Number of pulmonary emboli per lung slice (F) and the correlation between large embolic events and PE burden (G). All values are expressed as mean ± SEM; n = 6, 7, and 9 for WT, F5L/+, and F5L/L mice, respectively. *P < .05; **P < .01; ***P < .001, using a 1-way analysis of variance for panels A-D, Student t test for panel F, and linear regression for panel G.
In vivo mouse model of thrombus stability in WT, F5L/+,and F5L/Lmice. The change in thrombus size from baseline (A), number of total (B) and large (C) embolic events, and percentage of thrombus that embolized (D). (E) Representative emboli images found in the pulmonary arteries of WT and F5L/L mice. Number of pulmonary emboli per lung slice (F) and the correlation between large embolic events and PE burden (G). All values are expressed as mean ± SEM; n = 6, 7, and 9 for WT, F5L/+, and F5L/L mice, respectively. *P < .05; **P < .01; ***P < .001, using a 1-way analysis of variance for panels A-D, Student t test for panel F, and linear regression for panel G.
To determine whether the higher DVT turnover in WT mice led to increased PE burden, we quantified the number of pulmonary emboli per lung section in WT and F5L/L mice by histology (Figure 1E). PE burden was reduced in F5L/L mice compared with WT mice (Figure 1F). We have previously shown that the number of large embolic events correlate with PE burden.16 Here again, large embolic events correlated positively with PE burden in WT and F5L/L mice (Figure 1G). The increased thrombus size and reduced PE burden in F5L/L mice compared with WT (likely because of enhanced thrombus stability) provides a physiological/biochemical rationale for the FVL paradox. FVL carriers are more likely to present with symptomatic DVT from a small vessel injury. In contrast, noncarrriers have a higher incidence of embolization, which restricts the DVT to a subclinical size while at the same time leading to increased PE burden.
Several hypotheses have been proposed to account for the FVL paradox. One of the original hypotheses was that the presence of the FVL mutation would lead to fatal PE, resulting in a lower number of FVL subjects among those surviving with PE. However autopsy studies have shown no difference in the proportion of FVL among those with fatal PE compared with PE survivors or the general population.17,18 Other mechanisms such as thrombus location, the number of affected veins, thrombus growth, and density have all been investigated to explain the FVL paradox, but none were sufficient to explain it.13 A more recent hypothesis proposes that FVL may enhance local thrombin generation, thus intensifying the local inflammatory processes against the thrombus, as well as strengthening the clot structure by activation of thrombin-induced FXIII activity.13 However, other thrombophilic risk factors, such as antithrombin and protein C and S deficiencies, increase the risk for DVT as much as they increase the risk for PE, refuting this hypothesis. A second commonly proposed hypothesis involves an antifibrinolytic effect. Bajzar et al showed that in FVL heterozygotes there is an impaired TAFI-dependent profibrinolytic response to activated protein C.12 Parker et al later confirmed these results, and also showed that lysis of radiolabeled clots infused through the jugular vein and lodged in the lungs had less lysis in FVL homozygous mice compared with WT mice, supporting the hypothesis that FVL inhibits fibrinolysis.11 However, FVL carriers do not show an increased risk for postthrombotic syndrome,19,20 which would be expected if there was resistance to fibrinolysis. F5L mice had significantly increased F1.2 compared with WT (54.1 ± 7.3 ng/mL and 33.8 ± 2.1 ng/mL; P < .01). Therefore, we believe that after a small injury, a stable thrombus forms in FVL mice, with reduced embolization compared with WT. However, as there is significantly more thrombin generation in F5L mice (supported by the F1.2 data), this leads to thrombus growth and further stabilization, reducing embolization.
An increased rate of pulmonary microemboli in noncarriers compared with those with FVL has not been directly described. Indirectly, pulmonary microemboli are prevalent in acute respiratory distress syndrome, and those with FVL have a lower risk for death from acute respiratory distress syndrome than normal.21,22
In mice with the F5L/L mutation, the thrombus grows and stabilizes over time. Increased thrombus stability is inversely related to embolization and PE burden. This suggests that those with FVL have stable thrombus formation, leading to an increased incidence of symptomatic DVT and a decreased risk for PE.
Acknowledgments
The authors thank David Ginsburg’s laboratory for the FVL mice.
The study was partly funded by Canadian Venous Thromboembolism Clinical Trials and Outcomes Research.
Authorship
Contribution: S.A.S. performed all experiments, analyzed data and results, and wrote the manuscript; R.J.W. and P.L.G. supervised the research and conceived and designed the study; P.L.G. and R.J.W. contributed to the interpretation of data and critically edited the manuscript; and all authors have read and approved the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: P. L. Gross, Department of Medicine, Thrombosis and Atherosclerosis Research Institute, McMaster University, 237 Barton St East, Hamilton, ON L8L 2X2, Canada; e-mail: peter.gross@taari.ca.

