

Key Points
M2BPGi is increased in patients with liver graft-versus-host disease, especially in those at high risk for late NRM after allogeneic HSCT.
WFA+-M2BP–positive macrophages are found in liver graft-versus-host disease, supporting these cells as a responder of this glycoprotein.

Abstract
Macrophages play a crucial role in the pathogenesis of chronic graft-versus-host disease (cGVHD). We hypothesized that galectin-3, Mac-2 binding protein (M2BP), or Wisteria floribunda agglutinin (WFA)+-M2BP, called M2BP glycan isomer (M2BPGi), might contribute to macrophage activation, and fibrosis would be associated with cGVHD and nonrelapse mortality (NRM) in hematopoietic stem cell transplant (HSCT) recipients. Patients who underwent their first allogeneic HSCT and survived for >180 days without relapse were included. The predictive potential of the 3 markers for NRM was assessed using the discovery cohort (n = 55) and validation cohort 1 (n = 55). When we used the threshold determined by a receiver operating characteristics curve analysis in the discovery cohort, only M2BPGi at day +180 was significantly associated with a higher NRM in the discovery cohort (15.0% vs 0.0% at 5 years, P = .001) and in validation cohort 1 (34.0% vs 8.4% at 5 years, P = .014). This result was confirmed in validation cohort 2 (n = 50). M2BPGi was not increased in healthy individuals or in patients who received autologous HSCT. In the entire cohort (N = 110), M2BPGi was significantly related to liver cGVHD but not to other organ involvement. In multivariate analyses, M2BPGi was an independent risk factor for NRM. In immunofluorescence staining of autopsy cases, WFA+-M2BP–positive macrophages were found only in the liver sections with cGVHD. In conclusion, M2BPGi could be a promising predictor of late NRM after HSCT and was associated with liver involvement.
Introduction
Chronic graft-versus-host disease (cGVHD) is the most common long-term complication after allogeneic hematopoietic stem cell transplantation (HSCT)1,2 and leads to higher late nonrelapse mortality (NRM)3 and impaired quality of life in long-term survivors.4,5 Although many studies have identified several promising biomarkers for cGVHD,6 a suitable biomarker remains to be established for use in routine clinical practice.7,8
cGVHD is characterized by inflammation and fibrosis that compromise the function of multiple organs.9 Previous studies have demonstrated that macrophages play an important role in fibrosis.10 Macrophages express significant amounts of a β-galactoside–binding member of the lectin family, galectin-3 (GAL3), which drives inflammation, fibroblast proliferation, and collagen production.11 Meanwhile, Mac-2 binding protein (M2BP), known as GAL3 ligand, is also a possible candidate biomarker for fibrosis. This glycoprotein interacts with GAL3 and extracellular proteins, such as fibronectin.12 M2BP induces inflammatory cytokines, including interleukin-1 (IL-1), IL-6, and other cytokines from macrophages. Recently, Wisteria floribunda agglutinin (WFA)+-M2BP, which detects changes in the glycans on the surface of M2BP, has been introduced as a reliable glycobiomarker for liver fibrosis.13 WFA+-M2BP has recently been referred to as M2BP glycan isomer (M2BPGi).14
Here, we evaluated the plasma levels of GAL3, M2BP, and M2BPGi in 110 patients who received allogeneic HSCT and assessed their diagnostic potential for cGVHD and prognostic value for NRM.
Methods
Patient selection
The current study included 110 consecutive adult patients who received their first allogeneic HSCT at our center between January 2010 and December 2016 and survived for >180 days after HSCT without relapse. The diagnosis, severity, and response to treatment of cGVHD were based on the 2014 National Institutes of Health (NIH) consensus criteria.15,16
To evaluate the predictive potential of the 3 candidate biomarkers for NRM, the entire cohort was randomly divided into a discovery cohort (n = 55) and validation cohort 1 (n = 55). During this study period, 2 patients were excluded because they did not allow blood sample collection. The post hoc analysis in validation cohort 2 included 50 consecutive adult patients at our center who received their second or third allogeneic HSCT between January 2010 and December 2016 or their first allogeneic HSCT between January 2017 and June 2018 and survived for >180 days after HSCT without relapse. Their plasma samples were collected at around day +180 following transplantation and stored at −80°C until use. As controls, plasma samples were collected from 20 healthy adults and 11 patients who received autologous HSCT for malignant lymphoma (n = 5), multiple myeloma (n = 5), or acute promyelocytic leukemia (n = 1). The plasma levels of M2BPGi were indexed to the cutoff index (COI) values. See supplemental Materials for details on the measurement of the candidate biomarkers. Patients gave their written consent to allow blood sample collection in accordance with the Declaration of Helsinki. This study was approved by the Institutional Review Board of Jichi Medical University Saitama Medical Center.
Immunofluorescence staining
Formalin-fixed paraffin-embedded liver and lung tissues were obtained from 3 autopsy cases. Immunofluorescence staining was performed with the following primary antibodies: WFA lectin (Vector Laboratories, Burlingame, CA), rabbit anti-M2BP antibody (Proteintech, Rosemont, IL), and mouse anti-CD68 antibody and mouse anti–α-smooth muscle actin antibody (both from Dako, Glostrup, Denmark). We used streptavidin Alexa Fluor 488–conjugated antibody, Alexa Fluor 594 Chicken anti-Rabbit IgG antibody, and Alexa Fluor 594 Goat anti-Mouse IgG antibody (all from Invitrogen, Carlsbad, CA) as secondary antibodies. Tissues were cut 5-µm thick, deparaffinized, and rehydrated in a standard manner. To recover antigen in staining for WFA and M2BP, the sections were put into 100 mM citrate buffer, heated in a microwave for 5 minutes,17 and washed 3 times in 50 mM phosphate buffer, followed by blocking with Blocking One (Nacalai Tesque, Kyoto, Japan) for 10 minutes. Next, samples were incubated with primary antibodies overnight at 4°C and then incubated with streptavidin Alexa Fluor 488–conjugated antibody for 60 minutes and other secondary antibodies 3 times for 3 minutes. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI). WFA, M2BP, and DAPI triple-positive cells by immunofluorescence staining were defined as WFA+-M2BP–positive cells. We used a BZ-X700 Fluorescence Microscope to capture images, and images were processed and merged using BZ-X Analyzer software (both from Keyence, Osaka, Japan).
Statistical analysis
Dichotomous variables were compared using Fisher’s exact test. The Mann-Whitney U test and the Kruskal-Wallis test were used to compare continuous variables. The Jonckheere-Terpstra trend test was used to assess trends in ordered variables. The cumulative incidences of graft-versus-host disease (GVHD) and NRM were calculated using Gray’s method, considering relapse and death without GVHD as competing events for GVHD and relapse as a competing event for NRM. The Fine-Gray proportional hazards model was used in multivariate analyses for NRM, and nonsignificant covariates were removed in a stepwise manner. All P values were 2-sided, and significance was set at .05. All statistical analyses were performed with EZR version 1.37 (Jichi Medical University Saitama Medical Center), which is a graphical user interface for R (version 3.2.2; The R Foundation for Statistical Computing).18
Results
Patient characteristics
The baseline characteristics of the discovery cohort (n = 55), validation cohort 1 (n = 55), and validation cohort 2 (n = 50) are summarized in Table 1. The entire cohort (the discovery cohort and validation cohort 1, N = 110) included 1 hepatitis B virus (HBV) carrier (0.9%) and 1 hepatitis C virus (HCV) carrier (0.9%). GVHD prophylaxis consisted of cyclosporine or tacrolimus and short-term methotrexate. The median observation period of the survivors from sample collection was 1729 days (range, 111-2737). The overall survival rate at 5 years was 72.1% (95% confidence interval [CI], 61.9-79.9). The cumulative incidence of grade II-IV acute GVHD was 35.5% (95% CI, 26.9-44.8). The cumulative incidence of cGVHD at 5 years was 72.1% (95% CI, 62.5-79.6). The median onset of cGVHD was day +131 (range, 47-890).
Patient characteristics
| Discovery cohort (n = 55) | Validation cohort 1 (n = 55) | Validation cohort 2 (n = 50) | |
|---|---|---|---|
| Age at transplantation, y | |||
| Mean (range) | 48 (18-65) | 47 (18-66) | 46 (18-68) |
| Recipient/donor sex | |||
| Male/female | 13 (23.6) | 14 (25.5) | 11 (22.0) |
| Other combinations | 42 (76.4) | 41 (74.5) | 39 (78.0) |
| Underlying disease | |||
| Acute leukemia | 30 (54.5) | 40 (72.7) | 31 (62.0) |
| Malignant lymphoma | 5 (9.1) | 5 (9.1) | 6 (12.0) |
| MDS/MPN | 16 (29.1) | 5 (9.1) | 7 (14.0) |
| Aplastic anemia | 3 (5.5) | 5 (9.1) | 1 (2.0) |
| Chronic myeloid leukemia | 1 (1.8) | 0 (0) | 3 (6.0) |
| Other | 0 (0) | 0 (0) | 2 (4.0) |
| Disease risk* | |||
| Low risk | 51 (92.7) | 46 (83.6) | 46 (92.0) |
| High risk | 4 (7.3) | 9 (16.4) | 4 (8.0) |
| Performance status† | |||
| 0-1 | 54 (98.2) | 54 (98.2) | 48 (96.0) |
| 2-4 | 1 (1.8) | 1 (1.8) | 2 (4.0) |
| HCT-CI | |||
| 0-1 | 48 (87.3) | 47 (85.5) | 42 (84.0) |
| ≥2 | 7 (12.7) | 8 (14.5) | 8 (16.0) |
| Stem cell source | |||
| Bone marrow | 34 (61.8) | 31 (56.4) | 25 (50.0) |
| Peripheral blood stem cells | 16 (29.1) | 19 (34.5) | 24 (48.0) |
| Umbilical cord blood | 5 (9.1) | 5 (9.1) | 1 (2.0) |
| HLA | |||
| Match | 31 (56.4) | 25 (45.5) | 30 (60.0) |
| Mismatch | 24 (43.6) | 30 (54.5) | 20 (40.0) |
| Type of donor | |||
| Related | 16 (29.1) | 19 (34.5) | 21 (42.0) |
| Unrelated | 39 (70.9) | 36 (65.5) | 29 (58.0) |
| Conditioning regimen‡ | |||
| Myeloablative | 39 (70.9) | 39 (70.9) | 34 (68.0) |
| Reduced intensity | 16 (29.1) | 16 (29.1) | 16 (32.0) |
| GVHD prophylaxis | |||
| CSA + MTX | 47 (85.5) | 50 (90.9) | 44 (88.0) |
| TAC + MTX | 8 (14.5) | 5 (9.1) | 6(12.0) |
| Use of ATG/alemtuzumab | |||
| No | 49 (89.1) | 44 (80.0) | 37 (74.0) |
| Yes | 6 (10.9) | 11 (20.0) | 13 (26.0) |
| Prior acute GVHD§ | |||
| No | 16 (29.1) | 16 (29.1) | 9 (18.0) |
| Yes | 39 (70.9) | 39 (70.9) | 41 (82.0) |
| Prior cGVHD§ | |||
| No | 29 (52.7) | 33 (60.0) | 23 (46.0) |
| Yes | 26 (47.3) | 22 (40.0) | 27 (54.0) |
| Median days from HSCT to sample acquisition (range) | 179 (133-266) | 178 (168-274) | 181 (168-267) |
| Discovery cohort (n = 55) | Validation cohort 1 (n = 55) | Validation cohort 2 (n = 50) | |
|---|---|---|---|
| Age at transplantation, y | |||
| Mean (range) | 48 (18-65) | 47 (18-66) | 46 (18-68) |
| Recipient/donor sex | |||
| Male/female | 13 (23.6) | 14 (25.5) | 11 (22.0) |
| Other combinations | 42 (76.4) | 41 (74.5) | 39 (78.0) |
| Underlying disease | |||
| Acute leukemia | 30 (54.5) | 40 (72.7) | 31 (62.0) |
| Malignant lymphoma | 5 (9.1) | 5 (9.1) | 6 (12.0) |
| MDS/MPN | 16 (29.1) | 5 (9.1) | 7 (14.0) |
| Aplastic anemia | 3 (5.5) | 5 (9.1) | 1 (2.0) |
| Chronic myeloid leukemia | 1 (1.8) | 0 (0) | 3 (6.0) |
| Other | 0 (0) | 0 (0) | 2 (4.0) |
| Disease risk* | |||
| Low risk | 51 (92.7) | 46 (83.6) | 46 (92.0) |
| High risk | 4 (7.3) | 9 (16.4) | 4 (8.0) |
| Performance status† | |||
| 0-1 | 54 (98.2) | 54 (98.2) | 48 (96.0) |
| 2-4 | 1 (1.8) | 1 (1.8) | 2 (4.0) |
| HCT-CI | |||
| 0-1 | 48 (87.3) | 47 (85.5) | 42 (84.0) |
| ≥2 | 7 (12.7) | 8 (14.5) | 8 (16.0) |
| Stem cell source | |||
| Bone marrow | 34 (61.8) | 31 (56.4) | 25 (50.0) |
| Peripheral blood stem cells | 16 (29.1) | 19 (34.5) | 24 (48.0) |
| Umbilical cord blood | 5 (9.1) | 5 (9.1) | 1 (2.0) |
| HLA | |||
| Match | 31 (56.4) | 25 (45.5) | 30 (60.0) |
| Mismatch | 24 (43.6) | 30 (54.5) | 20 (40.0) |
| Type of donor | |||
| Related | 16 (29.1) | 19 (34.5) | 21 (42.0) |
| Unrelated | 39 (70.9) | 36 (65.5) | 29 (58.0) |
| Conditioning regimen‡ | |||
| Myeloablative | 39 (70.9) | 39 (70.9) | 34 (68.0) |
| Reduced intensity | 16 (29.1) | 16 (29.1) | 16 (32.0) |
| GVHD prophylaxis | |||
| CSA + MTX | 47 (85.5) | 50 (90.9) | 44 (88.0) |
| TAC + MTX | 8 (14.5) | 5 (9.1) | 6(12.0) |
| Use of ATG/alemtuzumab | |||
| No | 49 (89.1) | 44 (80.0) | 37 (74.0) |
| Yes | 6 (10.9) | 11 (20.0) | 13 (26.0) |
| Prior acute GVHD§ | |||
| No | 16 (29.1) | 16 (29.1) | 9 (18.0) |
| Yes | 39 (70.9) | 39 (70.9) | 41 (82.0) |
| Prior cGVHD§ | |||
| No | 29 (52.7) | 33 (60.0) | 23 (46.0) |
| Yes | 26 (47.3) | 22 (40.0) | 27 (54.0) |
| Median days from HSCT to sample acquisition (range) | 179 (133-266) | 178 (168-274) | 181 (168-267) |
Unless otherwise indicated, data are n (%).
ATG, anti-thymocyte globulin; CSA, cyclosporine; HCT-CI, hematopoietic cell transplantation-specific comorbidity index; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasm; MTX, methotrexate; TAC, tacrolimus.
Disease risk was categorized as low risk and high risk. High risk was defined as any hematological malignancy in noncomplete remission. Other conditions were considered low risk.
Performance status was evaluated by the Eastern Cooperative Oncology Group scale.
Conditioning intensity was classified based on criteria from the Center for International Blood and Marrow Transplant Research.
History of GVHD at sample acquisition.
Assessment of candidate biomarkers for NRM
We first investigated whether the 3 candidate biomarkers could predict NRM in the discovery cohort. According to the receiver operating characteristic (ROC) curves of the 3 candidates (Figure 1A), the thresholds of GAL3, M2BP, and M2BPGi at day +180 were 18, 70, and 1.5, respectively, with an area under the curve (AUC) of 0.64, 0.75, and 0.91, respectively. Using these thresholds, elevated plasma levels of M2BP and M2BPGi were significantly associated with higher NRM in the discovery cohort: 15.0% (95% CI, 4.5-31.4) vs 0.0% (95% CI, 0.0-0.0; P = .038) at 5 years from sample collection for M2BP and 23.3% (95% CI, 8.0-43.2) vs 0.0% (95% CI, 0.0-0.0; P = .001) at 5 years from sample collection for M2BPGi. In validation cohort 1, M2BP was not significantly associated with NRM: 29.8% (95% CI, 12.6-49.2) vs 13.4% (95% CI, 2.9-31.8; P = .101) at 5 years from sample collection. On the other hand, the adverse impact of M2BPGi on NRM was confirmed: 34.0% (95% CI, 15.7-53.3) vs 8.4% (95% CI, 1.3-24.5; P = .014) at 5 years from sample collection (Figure 1B). The ROC curve analysis of M2BPGi in validation cohort 1 showed an AUC of 0.82 (Figure 1C). To further confirm the impact of M2BPGi on NRM, we performed post hoc analysis using validation cohort 2. Although the median observation period of the survivors from sample collection in this cohort was relatively short (median, 531 days; range, 231-2891), M2BPGi was significantly associated with NRM: 35.3% (95% CI, 8.4-64.5) vs 4.0% (95% CI, 0.3-17.4; P = .045) at 2 years from sample collection (Figure 1B). Because M2BPGi was identified as a biomarker for liver fibrosis, we also performed ROC curve analysis of total bilirubin and alanine aminotransferase (ALT) in the entire cohort; AUC was 0.51, and 0.49, respectively (Figure 1D). Overall survival at 5 years from sample collection was significantly lower in the higher M2BPGi group: 57.8% (95% CI, 34.3-75.6) vs 87.5% (95% CI, 70.0-95.1; P = .017) in the discovery cohort. Overall survival at 5 years was not significantly different in validation cohort 1, probably because of the limited sample size: 60.7% (95% CI, 37.9-77.4) vs 76.2% (95% CI, 53.8-88.8; P = .164).

ROC curves in the discovery cohort, and NRM using the thresholds in the discovery cohort. (A) ROC curves of the 3 candidate biomarkers at day +180 for the prediction of NRM in the discovery cohort. ROC curves were used to define the thresholds of biomarkers for NRM by minimizing the distance from the upper left corner of the graph. The thresholds of GAL3, M2BP, and M2BPGi were 18, 70, and 1.5, and AUCs were 0.64, 0.75, and 0.91, respectively. (B) NRM in the discovery cohort, validation cohort 1, and validation cohort 2 stratified by M2BPGi at day +180 using the cutoff in the discovery cohort (thresholds of COI values: 1.5). NRM at 5 years from sample collection in the discovery cohort was 23.3% (95% CI, 8.0-43.2) for the group with higher M2BPGi and 0.0% (95% CI, 0.0-0.0) for the group with lower M2BPGi (P = .001). NRM at 5 years from sample collection in validation cohort 1 was 34.0% (95% CI, 15.7-53.3) for the group with higher M2BPGi and 8.4% (95% CI, 1.3-24.5) for the group with lower M2BPGi (P = .014). NRM at 2 years from sample collection in validation cohort 2 was 35.3% (95% CI, 8.4-64.5) for the group with higher M2BPGi and 4.0% (95% CI, 0.3-17.4) for the group with lower M2BPGi (P = .045). (C) ROC curve of M2BPGi at day +180 for the prediction of NRM in validation cohort 1 (AUC for M2BPGi, 0.82). (D) ROC curves for total bilirubin (left panel) and ALT (right panel) at day +180 for the prediction of NRM in the entire cohort (AUC for total bilirubin, 0.51; AUC for ALT, 0.49).
ROC curves in the discovery cohort, and NRM using the thresholds in the discovery cohort. (A) ROC curves of the 3 candidate biomarkers at day +180 for the prediction of NRM in the discovery cohort. ROC curves were used to define the thresholds of biomarkers for NRM by minimizing the distance from the upper left corner of the graph. The thresholds of GAL3, M2BP, and M2BPGi were 18, 70, and 1.5, and AUCs were 0.64, 0.75, and 0.91, respectively. (B) NRM in the discovery cohort, validation cohort 1, and validation cohort 2 stratified by M2BPGi at day +180 using the cutoff in the discovery cohort (thresholds of COI values: 1.5). NRM at 5 years from sample collection in the discovery cohort was 23.3% (95% CI, 8.0-43.2) for the group with higher M2BPGi and 0.0% (95% CI, 0.0-0.0) for the group with lower M2BPGi (P = .001). NRM at 5 years from sample collection in validation cohort 1 was 34.0% (95% CI, 15.7-53.3) for the group with higher M2BPGi and 8.4% (95% CI, 1.3-24.5) for the group with lower M2BPGi (P = .014). NRM at 2 years from sample collection in validation cohort 2 was 35.3% (95% CI, 8.4-64.5) for the group with higher M2BPGi and 4.0% (95% CI, 0.3-17.4) for the group with lower M2BPGi (P = .045). (C) ROC curve of M2BPGi at day +180 for the prediction of NRM in validation cohort 1 (AUC for M2BPGi, 0.82). (D) ROC curves for total bilirubin (left panel) and ALT (right panel) at day +180 for the prediction of NRM in the entire cohort (AUC for total bilirubin, 0.51; AUC for ALT, 0.49).
Among 30 patients in the entire cohort (discovery cohort and validation cohort 1) who died during the follow-up period, the cause of death, with the exception of primary disease relapse in the group with lower M2BPGi at day +180, was exclusively lung GVHD (n = 2) (Table 2). In contrast, two thirds of the patients with higher M2BPGi at day +180 died from GVHD or infection.
Causes of death
| Cause | Lower M2BPGi*(n = 11) | Higher M2BPGi (n = 19) |
|---|---|---|
| Relapse | 9 (81.8) | 4 (21.1) |
| Infection | 0 (0) | 3 (15.8) |
| Liver GVHD | 0 (0) | 3 (15.8) |
| Lung GVHD | 2 (18.2) | 4 (21.1) |
| GVHD except for liver and lung | 0 (0) | 2 (10.4) |
| Other | 0 (0) | 3 (15.8) |
| Cause | Lower M2BPGi*(n = 11) | Higher M2BPGi (n = 19) |
|---|---|---|
| Relapse | 9 (81.8) | 4 (21.1) |
| Infection | 0 (0) | 3 (15.8) |
| Liver GVHD | 0 (0) | 3 (15.8) |
| Lung GVHD | 2 (18.2) | 4 (21.1) |
| GVHD except for liver and lung | 0 (0) | 2 (10.4) |
| Other | 0 (0) | 3 (15.8) |
Data are n (%).
COI values < 1.5.
Impact of M2BPGi on NRM at day +90 and day +365
To evaluate the prognostic impact of M2BPGi at different time points, samples at day +90 (n = 110) and day +365 (n = 100) were used from the discovery cohort and validation cohort 1. Samples were actually obtained at a median of day +91 (range, 59-112) and day +363 (range, 259-527), respectively. At the earlier time point, at around day +90, NRM did not differ significantly according to the M2BPGi value in validation cohort 1 (discovery cohort, 20.9% vs 2.9% at 5 years, P = .016; validation cohort 1, 24.2% vs 16.1% at 5 years, P = .690; supplemental Figure 1A). In contrast, higher M2BPGi at around day +365 remained a significant risk factor for NRM in both cohorts (discovery cohort, 17.6% vs 0.0% at 5 years, P = .007; validation cohort 1, 40.8% vs 6.1% at 5 years, P = .008; supplemental Figure 1B).
Characteristics of patients with elevated M2BPGi
The discovery cohort and validation cohort 1 were combined to evaluate the clinical features with regard to the M2BPGi value. The levels of M2BPGi in the group with allogeneic HSCT at day +180 were compared with those in patients who underwent autologous HSCT or healthy controls. The median day of sample collection in patients with autologous HSCT was day +187 (range, 111-335). The COI value of M2BPGi in the group with allogeneic HSCT (median COI, 1.31; range, 0.21-16.25) was higher than that in the group with autologous HSCT (median COI, 0.82; range, 0.26-1.48; P = .014) or in the healthy controls (median COI, 0.40; range, 0.20-0.69; P < .001; Figure 2A). None of the patients with autologous HSCT or healthy controls showed a COI for M2BPGi ≥ 1.5. These results suggested that M2BPGi might be increased in the network of allogeneic immune responses.
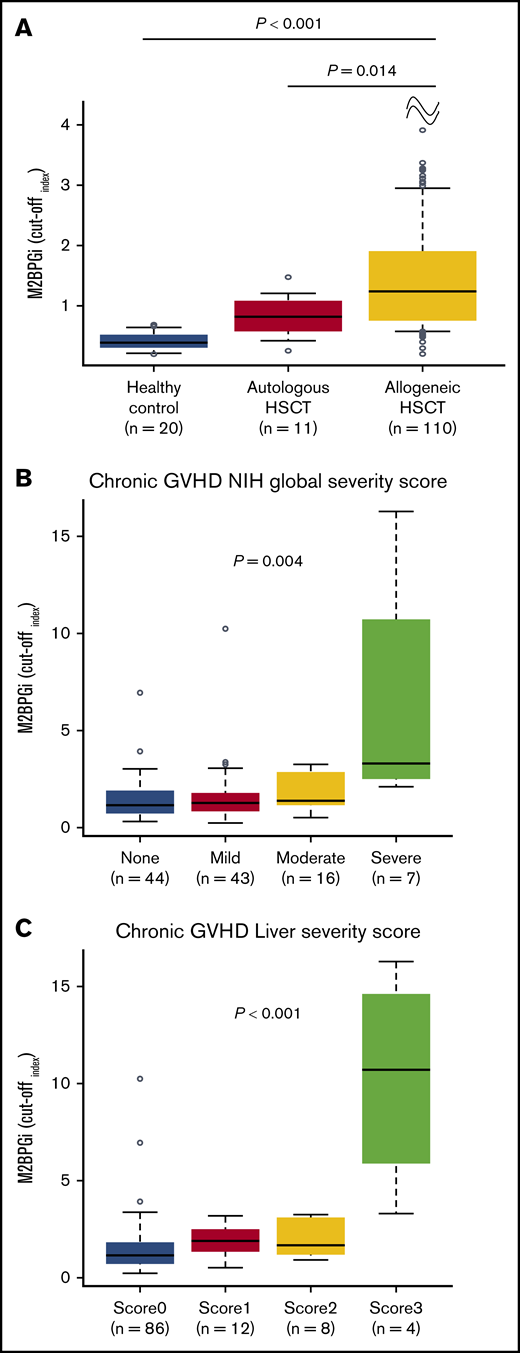
M2BPGi values and relationship between COI values for M2BPGi and GVHD severity score. (A) COI values for M2BPGi in the healthy control group (n = 20) and in patients who underwent autologous HSCT (n = 11) and allogeneic HSCT (n = 110). The group with allogeneic HSCT consisted of the discovery cohort and validation cohort 1. Samples with autologous HSCT and allogeneic HSCT were collected at day +180. Data are shown as box-and-whisker plots that indicate the 90th and 10th percentiles. Horizontal black lines represent the median values, and circles indicate outliers. P values compare the group with allogeneic HSCT with the healthy control group or with the group with autologous HSCT. The Mann-Whitney U test was used to compare the data. Association between M2BPGi at day +180 and NIH global severity score (B) or liver severity score (C) in the entire cohort. The NIH global severity score and liver severity score were evaluated at sample collection. Data are shown as box-and-whisker plots that indicate the 90th and 10th percentiles. Horizontal black lines represent median values, and circles indicate outliers. The Jonckheere-Terpstra test was used to evaluate the association between M2BPGi and the NIH global severity score or liver severity score.
M2BPGi values and relationship between COI values for M2BPGi and GVHD severity score. (A) COI values for M2BPGi in the healthy control group (n = 20) and in patients who underwent autologous HSCT (n = 11) and allogeneic HSCT (n = 110). The group with allogeneic HSCT consisted of the discovery cohort and validation cohort 1. Samples with autologous HSCT and allogeneic HSCT were collected at day +180. Data are shown as box-and-whisker plots that indicate the 90th and 10th percentiles. Horizontal black lines represent the median values, and circles indicate outliers. P values compare the group with allogeneic HSCT with the healthy control group or with the group with autologous HSCT. The Mann-Whitney U test was used to compare the data. Association between M2BPGi at day +180 and NIH global severity score (B) or liver severity score (C) in the entire cohort. The NIH global severity score and liver severity score were evaluated at sample collection. Data are shown as box-and-whisker plots that indicate the 90th and 10th percentiles. Horizontal black lines represent median values, and circles indicate outliers. The Jonckheere-Terpstra test was used to evaluate the association between M2BPGi and the NIH global severity score or liver severity score.
Next, the associations between the M2BPGi value and organ involvement or severity of cGVHD at day +180 were assessed. Liver cGVHD was significantly associated with higher M2BPGi (P = .025), but the involvement of other organs was not significantly associated with an increased level of M2BPGi (supplemental Figure 2). The levels of M2BPGi were significantly associated with the NIH global severity score (P = .004; Figure 2B) and liver severity score (P < .001; Figure 2C) at day +180. Consequently, to determine the true impact of M2BPGi for NRM, we performed multivariate analyses that included the possible confounders at HSCT and at sample collection. The possible confounders at HSCT were age (<50 vs ≥50 years), sex (female to male vs others), hematopoietic cell transplantation-specific comorbidity index (HCT-CI; 0-1 vs ≥2), HLA compatibility (match vs mismatch), and use of anti-thymocyte globulin (ATG)/alemtuzumab. The possible confounders at sample collection included the NIH global severity score (0-2 vs 3) or liver severity score (0-2 vs 3) and the platelet count (<100 000 per microliter vs ≥100 000 per microliter), which is a well-known prognostic factor in GVHD.19,20 Because the NIH global severity score and liver severity score were highly correlated, each covariate was subjected to a separate multivariate analysis. As a result, higher M2BPGi remained an independent risk factor for NRM (hazard ratio [HR], 9.20; 95% CI, 1.87-45.26; P = .006; HR, 8.52; 95% CI, 1.67-43.51; P = .010; Table 3).
Risk factors for NRM by univariate and multivariate analyses
| Univariate analysis | Multivariate analysis* | Multivariate analysis* | ||||
|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | |
| M2BPGi | ||||||
| COI < 1.5 | 1 | Ref | 1 | Ref | 1 | Ref |
| COI ≥ 1.5 | 11.27 (2.61-48.67) | 0.001 | 9.20 (1.87-45.26) | 0.006 | 8.52 (1.67-43.51) | 0.010 |
| Thrombocytopenia† | ||||||
| No | 1 | Ref | 1 | Ref | ||
| Yes | 3.40 (1.25-9.26) | 0.017 | 3.60 (1.03-12.63) | 0.045 | ||
| NIH global severity score‡ | ||||||
| 0/1/2 | 1 | Ref | 1 | Ref | ||
| 3 | 13.8 (4.50-42.06) | <0.001 | 4.46 (1.44-13.84) | 0.010 | ||
| Liver severity score§ | ||||||
| 0/1/2 | 1 | Ref | 1 | Ref | ||
| 3 | 12.2 (3.40-43.53) | <0.001 | 9.24 (2.06-41.40) | 0.004 | ||
| HCT-CI | ||||||
| 0-1 | 1 | Ref | 1 | Ref | 1 | Ref |
| ≥2 | 3.68 (1.25-10.89) | 0.018 | 3.17 (1.16-8.69) | 0.025 | 3.48 (1.12-10.87) | 0.032 |
| Univariate analysis | Multivariate analysis* | Multivariate analysis* | ||||
|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | |
| M2BPGi | ||||||
| COI < 1.5 | 1 | Ref | 1 | Ref | 1 | Ref |
| COI ≥ 1.5 | 11.27 (2.61-48.67) | 0.001 | 9.20 (1.87-45.26) | 0.006 | 8.52 (1.67-43.51) | 0.010 |
| Thrombocytopenia† | ||||||
| No | 1 | Ref | 1 | Ref | ||
| Yes | 3.40 (1.25-9.26) | 0.017 | 3.60 (1.03-12.63) | 0.045 | ||
| NIH global severity score‡ | ||||||
| 0/1/2 | 1 | Ref | 1 | Ref | ||
| 3 | 13.8 (4.50-42.06) | <0.001 | 4.46 (1.44-13.84) | 0.010 | ||
| Liver severity score§ | ||||||
| 0/1/2 | 1 | Ref | 1 | Ref | ||
| 3 | 12.2 (3.40-43.53) | <0.001 | 9.24 (2.06-41.40) | 0.004 | ||
| HCT-CI | ||||||
| 0-1 | 1 | Ref | 1 | Ref | 1 | Ref |
| ≥2 | 3.68 (1.25-10.89) | 0.018 | 3.17 (1.16-8.69) | 0.025 | 3.48 (1.12-10.87) | 0.032 |
Because the NIH global severity score and liver severity score were highly correlated, each covariate was subjected to a separate multivariate analysis.
Ref, reference.
Other variables that were included in the multivariate analysis were age (<50 vs ≥50 years), sex (female to male vs others), HCT-CI (0-1 vs ≥2), HLA compatibility (match vs mismatch), and use of ATG/alemtuzumab.
Thrombocytopenia was defined as <100 000 platelets per microliter at sample collection.
NIH global severity score was evaluated at sample collection.
Liver severity score was evaluated at sample collection.
In addition, M2BPGi was significantly associated with higher NRM in patients with liver GVHD or without liver GVHD at day +180 (with liver GVHD, 31.2% vs 0.0% at 5 years; P < .001; without liver GVHD, 27.6% vs 4.6% at 5 years; P < .001; supplemental Figure 3).
Immunofluorescence staining in 3 autopsy cases
M2BPGi was significantly associated with liver GVHD, whereas the cause of NRM in the group with lower M2BPGi (COI < 1.5) was lung GVHD. We hypothesized that GVHD might lead to an increased number of WFA+-M2BP–positive cells in the liver but not the lung. Therefore, immunofluorescence staining of liver and lung samples was performed in 3 autopsy cases (Table 4). See supplemental Materials for detailed patient clinical courses. Briefly, Patient 1, who was an HBV carrier, developed treatment-refractory liver GVHD without lung GVHD. The HBV DNA concentration in peripheral blood remained at an undetectable level until death under entecavir prophylaxis. Patient 2 developed liver GVHD and achieved a partial response after treatment but died of bronchiolitis obliterans (BO). Patient 3 developed BO without liver GVHD.
Patient characteristics of 3 autopsy cases
| Patient | Age, y | Primary disease | Donor type | Stem cell source | Conditioning | Hepatitis infections | Acute GVHD | Onset of liver GVHD, d | Onset of lung cGVHD, d | Other cGVHD | Death, d | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 44 | AML | MRD | PB | Cy/TBI | HBsAg(+) | None | +94 | None | Skin | +887 | Liver failure |
| 2 | 66 | CMMoL | MRD | PB | Flu/Bu | None | Grade I | +123 | +186 | Skin | +372 | BO |
| 3 | 58 | FL | Haplo | PB | Flu/Mel/TBI | None | None | None | +444 | None | +618 | Multiple infections |
| Patient | Age, y | Primary disease | Donor type | Stem cell source | Conditioning | Hepatitis infections | Acute GVHD | Onset of liver GVHD, d | Onset of lung cGVHD, d | Other cGVHD | Death, d | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 44 | AML | MRD | PB | Cy/TBI | HBsAg(+) | None | +94 | None | Skin | +887 | Liver failure |
| 2 | 66 | CMMoL | MRD | PB | Flu/Bu | None | Grade I | +123 | +186 | Skin | +372 | BO |
| 3 | 58 | FL | Haplo | PB | Flu/Mel/TBI | None | None | None | +444 | None | +618 | Multiple infections |
AML, acute myeloid leukemia; Bu, busulfan; CMMoL, chronic myelomonocytic leukemia; Cy, cyclophosphamide; FL, follicular lymphoma; Flu, fludarabine; Haplo, haploidentical donor; Mel, melphalan; HBsAg, hepatitis B surface antigen; MRD, matched related donor; PB, peripheral blood stem cells; TBI, total body irradiation.
First, we evaluated tissue fibrosis in the liver and lung in each patient. As expected, α-smooth muscle actin immunofluorescence staining showed significantly more fibrotic areas in the involved sites of liver GVHD (Patients 1 and 2) and lung GVHD (Patients 2 and 3) (supplemental Figure 4). Liver fibrosis score based on the METAVIR staging system in Patients 1, 2, and 3 was F4, F2, and F0, respectively. We then determined the localization of WFA+-M2BP–positive cells in the liver and lung sections. WFA, M2BP, and DAPI triple-positive cells by immunofluorescence staining were defined as WFA+-M2BP–positive cells. Immunofluorescence staining of liver sections showed that WFA+-M2BP–positive cells were present in Patients 1 and 2 (Figure 3A). No WFA+-M2BP–positive cells were identified in Patient 3, who did not have liver GVHD. The number of WFA+-M2BP–positive cells in Patient 1 with active liver GVHD was significantly higher than that in Patient 2, who achieved a partial response following an increase in cyclosporine (P < .001) (Figure 3B). In contrast, immunofluorescence staining of lung sections revealed no WFA+-M2BP–positive cells in patients with BO regardless of fibrotic changes. These results suggested that COI values of M2BPGi increased in association with the number of WFA+-M2BP–positive cells in the liver, which reflected GVHD activity for the liver but not for the lung. In addition, immunofluorescence staining of serial sections in Patient 1 revealed that WFA+-M2BP–positive cells in the liver were also positive for the macrophage marker CD68 (Figure 3C-D). WFA+-M2BP–positive cells in liver sections from Patient 2 were also positive for CD68. On the other hand, despite the lack of WFA+-M2BP-positive cells in the lung, CD68+ macrophages were observed in the lungs of all patients (supplemental Figure 5).
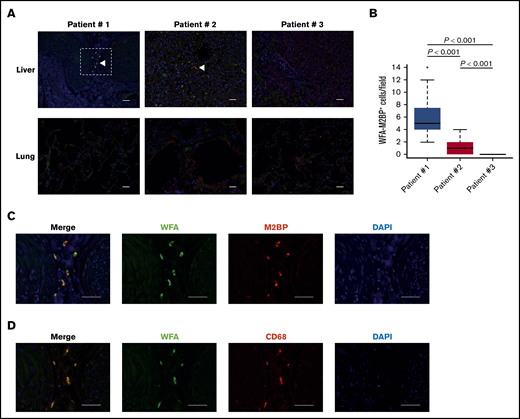
Identification of WFA+-M2BP–positive cells in immunofluorescent images. (A) Representative immunofluorescent images of WFA (green), M2BP (red), and DAPI (blue). Liver (upper panels) and lung (lower panels) sections from Patients 1, 2, and 3 are shown. WFA+-M2BP–positive cells in liver sections from Patients 1 and 2. Arrowheads point to examples of WFA+-M2BP–positive cells. Scale bars, 50 μm. (B) Quantification of WFA+-M2BP–positive cells in liver sections. The total number of WFA+-M2BP–positive cells was counted in 20 fields of view for each patient at original magnification ×200. Data are shown as box-and-whisker plots that indicate the 90th and 10th percentiles. Horizontal black lines represent median values, and circles indicate outliers. The Kruskal-Wallis nonparametric comparison with the Bonferroni adjustment was used for multiple comparisons. (C-D) Higher-magnification views of the area outlined by the dashed box in panel A. Scale bars, 50 μm. Representative immunofluorescent images of WFA (green), M2BP (red), and DAPI (blue) (C) and WFA (green), CD68 (red), and DAPI (blue) (D).
Identification of WFA+-M2BP–positive cells in immunofluorescent images. (A) Representative immunofluorescent images of WFA (green), M2BP (red), and DAPI (blue). Liver (upper panels) and lung (lower panels) sections from Patients 1, 2, and 3 are shown. WFA+-M2BP–positive cells in liver sections from Patients 1 and 2. Arrowheads point to examples of WFA+-M2BP–positive cells. Scale bars, 50 μm. (B) Quantification of WFA+-M2BP–positive cells in liver sections. The total number of WFA+-M2BP–positive cells was counted in 20 fields of view for each patient at original magnification ×200. Data are shown as box-and-whisker plots that indicate the 90th and 10th percentiles. Horizontal black lines represent median values, and circles indicate outliers. The Kruskal-Wallis nonparametric comparison with the Bonferroni adjustment was used for multiple comparisons. (C-D) Higher-magnification views of the area outlined by the dashed box in panel A. Scale bars, 50 μm. Representative immunofluorescent images of WFA (green), M2BP (red), and DAPI (blue) (C) and WFA (green), CD68 (red), and DAPI (blue) (D).
Discussion
Here, we found that the M2BPGi value measured at approximately day 180 was a good predictor of late NRM after allogeneic HSCT and was significantly correlated with liver GVHD. In contrast, M2BPGi was not associated with lung GVHD or an increased risk for NRM early after HSCT.
The development of biomarkers for cGVHD, especially specific to each GVHD target organ, is an important challenge, because several organs may be involved, and the manifestations are heterogeneous. Over the last decade, nucleic acids and proteins have primarily been analyzed as candidate biomarkers for GVHD.6 Meanwhile, glycans on the surface of most proteins are attractive candidates for biomarker research, because their structures in vivo are altered under environmental changes resulting from various diseases. For example, AFP-L3 has been approved as a glycobiomarker for hepatocellular carcinoma by the US Food and Drug Administration. A novel glycan-profiling technology using lectin microarrays allows a rapid analysis of complicated features of glycans.21 These glycoproteomics technologies have identified M2BPGi as a novel glycobiomarker for liver fibrosis due to HCV infection.13 Briefly, M2BP oligomerizes in large ring structures covered with N-glycans and undergoes N-glycosylation. These glycans are altered during the progression of liver fibrosis. Novel glycoproteomics techniques, using a lectin microarray system, identified the optimal lectin, WFA, to detect the changes in the N-glycosylation of M2BP.13 M2BPGi represents the quantification of WFA+-M2BP by sandwich immunoassay using WFA and anti-M2BP antibody.
In immunofluorescence staining in autopsy cases, WFA+-M2BP–positive cells with the macrophage marker CD68 were exclusively found in liver sections with GVHD. Immunofluorescence staining also showed that the number of WFA+-M2BP–positive cells was increased in active liver GVHD (Patient 1) compared with inactive liver GVHD after treatment (Patient 2). Similarly, plasma M2BPGi was associated with the liver severity score at the time of sample collection. These results indicated that WFA+-M2BP–positive macrophages might be involved in the development of liver GVHD, leading to higher NRM. A previous study of liver fibrosis demonstrated that WFA+-M2BP was produced from hepatic stellate cells, and WFA+-M2BP secreted from hepatic cells was transferred to the cell surface of macrophages.17 In addition, WFA+-M2BP was shown to have a profound impact on the activation of macrophages and the progression of fibrosis. Taken together, these results suggest that WFA+-M2BP might contribute to liver fibrosis in cGVHD, as well as HCV-induced cirrhosis. However, WFA+-M2BP–positive cells were not identified in lung tissue with a significant fibrotic change, despite macrophage infiltration. Because macrophages play an important role, even in a murine model of lung GVHD,22 different pathways might be essential to induce macrophage activation and fibrosis in the lung. The cross talk mechanisms involved in WFA+-M2BP–induced macrophage activation and fibrosis in each GVHD target organ should be elucidated in future studies.
Although the cause of NRM in the group with lower M2BPGi (COI < 1.5) was lung GVHD, various causes of NRM were observed in the group with higher M2BPGi. One possible explanation is that enhanced immunosuppressive therapy for liver GVHD resulted in various infections or other complications. Actually, Inamoto et al showed that severe liver GVHD was associated with an increased risk for mortality.23 However, because higher M2BPGi was associated with an increased risk for NRM in the analysis limited to patients without liver GVHD at day +180, other mechanisms independent from liver GVHD might contribute to higher NRM.
Our study also confirmed the prognostic significance of M2BPGi at day +365 but not at day +90. Previous studies demonstrated that M2BPGi reflected various forms of liver damage, even in the absence of fibrosis.24,25 In the early phase after allogeneic HSCT, acute liver injuries could occur as a result of various causes, such as acute liver GVHD, viral hepatitis, veno-occlusive disease, and drug exposure. Therefore, the implications of elevated M2BPGi might be different depending on the timing after HSCT.
This study has several limitations. First, although M2BPGi showed significant prognostic value in the 3 cohorts, this should be confirmed in a larger independent validation cohort.7 In addition, only samples at fixed time points were available in this study. M2BPGi is a possible biomarker for response to treatment, because its plasma concentration and the number of WFA+-M2BP–positive cells were associated with liver GVHD and NRM. To determine the impact of M2BPGi as a biomarker of the therapeutic response, M2BPGi should be measured before and after the initiation of therapy. Second, candidate biomarkers specific to target organs should be evaluated in patients with a similar phenotype. Indeed, our study suggested that the progression of lung fibrosis in GVHD was independent of WFA+-M2BP, in contrast to liver GVHD. Third, autopsy Patient 1 was an HBV carrier. Several studies have not found a relationship between liver GVHD and HBV infection.26,27 Although the HBV DNA concentration in peripheral blood remained at an undetectable level under the use of entecavir after HSCT, it is difficult to deny the possibility that this liver GVHD and liver failure were attributable to HBV infection.
In conclusion, M2BPGi could be a promising biomarker that predicts late NRM after allogeneic HSCT. M2BPGi was also associated with the severity of cGVHD and liver involvement. Moreover, WFA+-M2BP–positive macrophages might contribute to the pathogenesis of liver cGVHD. Importantly, this study indicated that novel glycan-profiling technologies could be powerful tools for identifying potential biomarkers for GVHD in the near future.
Acknowledgment
The authors thank all of the clinicians at our center who helped to obtain data for this study.
Authorship
Contribution: Y.A. designed the study, performed experiments, collected data, analyzed data, and wrote the first draft of manuscript; H.N. designed the study, advised on methods, and wrote the manuscript; K.K., M.K., S. Kawamura, J.T., N.Y., Y.M., K.Y., A.G., A.T., M.T., S.-i.K., and S. Kako collected data; and Y.K. organized the project and reviewed and revised the manuscript.
Conflict-of-interest disclosures: The authors declare no competing financial interests.
Correspondence: Yoshinobu Kanda, Division of Hematology, Jichi Medical University Saitama Medical Center, 1-847 Amanuma-cho, Omiya-ku, Saitama City, Saitama 330-8503, Japan; e-mail: ycanda-tky@umin.ac.jp.

