

Key Points
CD34+CD7+ as well as CD34−CD7+ cells from SR1-expanded CD34+ HSPCs are effective thymus-reconstituting cells in vivo.
CD7+ cells derived from SR1-expanded CD34+ HSPCs generate functional and polyclonal T-cell repertoires in vivo.

Abstract
Broader clinical application of umbilical cord blood (UCB), as a source of hematopoietic stem/progenitor cells (HSPCs), is limited by low CD34+ and T-cell numbers, contributing to slow lymphohematopoietic recovery, infection, and relapse. Studies have evaluated the safety, feasibility, and expedited neutrophil recovery associated with the transplantation of CD34+ HSPCs from ex vivo expansion cultures using the aryl hydrocarbon receptor antagonist StemRegenin-1 (SR1). In a phase 1/2 study of 17 patients who received combined SR1-expanded and unexpanded UCB units, a considerable advantage for enhancing T-cell chimerism was not observed. We previously showed that progenitor T (proT) cells generated in vitro from HSPCs accelerated T-cell reconstitution and restored immunity after hematopoietic stem cell transplantation (HSCT). To expedite immune recovery, we hypothesized that SR1-expanded HSPCs together with proT cells could overcome the known T-cell immune deficiency that occurs post-HSCT. Here, we show that SR1-expanded UCB can induce >250-fold expansion of CD34+ HSPCs, which can generate large numbers of proT cells upon in vitro differentiation. When compared with nonexpanded naive proT cells, SR1 proT cells also showed effective thymus-seeding and peripheral T-cell functional capabilities in vivo despite having an altered phenotype. In a competitive transfer approach, both naive and SR1 proT cells showed comparable thymus-engrafting capacities. Single-cell RNA sequencing of peripheral CD3+ T cells from mice injected with either naive or SR1 proT cells revealed functional subsets of T cells with polyclonal T-cell receptor repertoires. Our findings support the use of SR1-expanded UCB grafts combined with proT-cell generation for decreasing T-cell immunodeficiency post-HSCT.
Introduction
T cells are critical mediators of antiviral and antifungal immunities and are key players in the prevention of relapse after hematopoietic stem cell transplantation (HSCT).1 However, there is a lack of transferred adoptive immunity and incomplete reconstitution of a polyclonal T-cell repertoire in the host during HSCT, as a result of both a conditioning-induced defective thymic microenvironment and decreased production of progenitor T (proT) cells. Our group and others have previously reported the use of the OP9-DL1 cell coculture system for ex vivo generation of proT cells from multiple stem cell sources, including from human umbilical cord blood (UCB).1,,,,,,,-9 Adoptive transfer of human proT cells together with human hematopoietic stem/progenitor cells (HSPCs) allowed for enhanced HSPC-derived T-cell reconstitution in a preclinical model of HSCT.6,8 Thus, using in vitro–derived proT cells from UCB HSPCs could provide an adoptive cell therapy to overcome immunodeficiency after HSCT,10 if sufficient proT cell numbers could be generated in vitro from a single UCB unit.
There have been several efforts to increase the absolute number of HSPCs in UCB transplantation through transplanting 2 UCB units at 1 time11 or through ex vivo expansion cultures using cytokines,12,,,,-17 recombinant Notch ligands,18,19 or small molecules.20,21 StemRegenin-1 (SR1), an aryl hydrocarbon receptor antagonist, was the first compound identified in an unbiased screen for its ability to promote the expansion of CD34+ HSPCs in combination with cytokines.21 In a phase 1/2 trial of SR1-expanded UCB units, SR1 produced a median 330-fold increase in CD34+ HSPCs, led to engraftment in 17 of 17 patients, and significantly expedited neutrophil and platelet recovery compared with patients treated with unmanipulated UCB (naive UCB).22 Notably, SR1-expanded HSPCs were safe for transplantation.11,22 Although promising, there was no difference observed in T-cell reconstitution 360 days after transplantation of SR1-expanded HSPCs compared with naive HSPCs in this study. Therefore, the transfer of proT cells during HSCT using SR1 UCB has important implications for immune reconstitution and remains to be explored.
Here, we extend our previous studies and show that SR1 expansion of CD34+ UCB cells generates >250-fold more HSPCs, thus leading to more proT cells compared with naive UCB on OP9-DL1 cells. These proT cells had a slightly different developmental phenotype and were capable of thymus reconstitution in an immunodeficient mouse model. Upon competitive reconstitution of naive and SR1-expanded proT cells, both subsets engrafted the thymus at comparable frequencies. Furthermore, mice injected with either naive or SR1 proT cells generated functional subsets of T cells bearing diverse and polyclonal T-cell receptor (TCR) repertoires. Our findings provide support for the use of SR1-expanded UCB grafts, combined with OP9-DL1–based differentiation of proT cells, as a novel allogeneic strategy for promoting T-cell recovery during periods of immunodeficiency after HSCT.
Methods
UCB samples
Human UCB samples were obtained, and HSPC-containing fractions were purified using CD34 progenitor cell isolation kits (Miltenyi Biotec) following manufacturer protocol as previously described,5 in accordance with approved guidelines established by the Research Ethics Board of Sunnybrook Health Sciences Centre.
Mice
NOD.cg-PrkdcscidIL2rgtm/Wjl/Sz (NOD/SCID/γcnull [NSG]) mice were purchased from Jackson Laboratory (Bar Harbor, ME) and housed and bred in a pathogen-free facility. All animal procedures were approved by the Sunnybrook Health Sciences Centre Animal Care Committee.
SR1 expansion
All expansion experiments were performed in HSC expansion media, consisting of StemSpan Serum-Free Expansion Medium (StemCell Technologies) supplemented with 1× antibiotics, 0.75 μM of SR1 (StemCell Technologies), and the following recombinant human cytokines at 50 ng/mL: thrombopoietin, interleukin-6 (IL-6), Flt-3 ligand (Flt-3L), and stem cell factor (SCF; R&D Systems). CD34+ HSPCs were resuspended in HSC expansion medium at 5 × 103 cells per mL and cultured in culture flasks. Cells were transferred to larger flasks with fresh medium as needed to keep the cell density between 3 × 105 and 1 × 106 cells per mL.21 Cells were cultured at 37°C in 5% CO2. After expansion, cells were frozen (5 × 106 to 10 × 106/mL) in Cryostor10 freezing medium (StemCell Technologies).
Progenitor T-cell cocultures with OP9-DL1 cells
OP9-DL1 cells23 were maintained in α-MEM medium supplemented with 20% fetal bovine serum (Gibco), plus 100 U/mL of penicillin/streptomycin (OP9-media). OP9-DL1 cells were γ irradiated (100 Gy) and seeded onto tissue culture flasks. SR1 UCB and naive UCB were thawed as previously described for naive HSPCs24 and seeded at a 2:1 ratio to OP9-DL1 cells in OP9 media supplemented with recombinant human cytokines Flt-3L (5 ng/mL), IL-7 (5 ng/mL), and SCF (50 ng/mL; Miltenyi). Every 5 days, cocultures were transferred onto a fresh confluent monolayer of irradiated OP9-DL1 cells.24 Cocultures were carried out for 13 to 14 days.
Engraftment of progenitor T cells in NSG mice
CD34+CD7+, CD34−CD7+, or bulk CD7+ cells were sorted from day 13 to 14 naive or SR1 UCB/OP9-DL1 cultures, resuspended in a mixture containing recombinant human IL-7 (0.5 μg) and IL-7 antibody M25 (2.5 μg; provided by C. Surh, Scripps Research Institute, La Jolla, CA), and injected intrahepatically (30 μL per mouse) into nonirradiated 2- to 5-day-old NSG neonates as previously described.5,25 For intracellular cytokine staining, splenocytes were harvested from NSG mice 10 to 12 weeks after intrahepatic injection of SR1 CD7+ cells. Cells were incubated for 6 hours with 50 ng/mL of phorbol 12-myristate 13-acetate (Sigma Aldrich), 500 ng/mL of ionomycin (Sigma Aldrich), and 3 μg/mL of brefeldin A (ThermoFisher). For TCR-based stimulation, CD3+ cells were sorted and activated with Immunocult Human CD3/CD28 T Cell Activator in ImmunoCult-XF T Cell Expansion Medium (StemCell Technologies) for 3 days, following manufacturer protocol. Stimulated cells were washed poststimulation and stained for intracellular cytokines.
Naive/SR1 CD7+ cell coinjection
Naive UCB CD34+ cells incubated in X-VIVO10 serum-free hematopoietic cell media (Lonza, Basel, Switzerland) containing thrombopoietin (10 ng/mL), Flt-3L (100 ng/mL), SCF (100 ng/mL), and IL-3 (30 ng/mL). Twenty four hours later, 1 × 105 CD34+ cells were added to retronectin-coated (20 μg/mL; Clontech) plates with lentiviral vector at a multiplicity of infection of 50 and incubated for 24 hours. Naive ZsGreen+ HSPCs were sorted and placed into OP9-DL1 coculture in parallel with untransduced SR1 HSPCs. Cocultures were sorted for CD7+ proT cells, and 3 × 105 cells of each subset were coinjected into NSG neonates.
Single-cell RNA sequencing
NSG mice were injected with 5 × 105 to 7 × 105 sorted CD7+ proT cells from either day-14 naive or SR1 UCB/OP9-DL1 cocultures. Ten weeks postinjection, splenocytes were harvested, and flow cytometrically sorted CD3+ T cells (pooled from n = 3 mice) were RNA sequenced at single-cell resolution using 10X Genomics. Data were analyzed using R software with the package Seurat.26 Each sample, from a total of 1359 naive and 683 SR1 cells, was first filtered to remove cells with low gene counts (naive, n = 92;SR1, n = 37) that arise from aborted sequencing, and gene expression was normalized between cells. Afterward, variable expression of genes was determined. Naive and SR1 samples were then merged and aligned; dimensions for t-distributed stochastic neighbor embedding were calculated to identify unique cell clusters, and the differential expression of genes between cell clusters was determined. Cell subsetting was performed to identify and analyze CD4 and CD8A-expressing T-cell populations.26 Clonotype, Vβ, and complementarity determining region 3 (CDR3) gene analyses were performed using R software with the package Immunarch (Immunomind) after filtering out clonotypes with ambiguous gene alignments. The data discussed in this report have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus27 and are accessible through GEO Series accession #GSE135929.
Flow cytometry
Cell suspensions were stained for surface markers (supplemental Table 1) and analyzed with an LSR-II cytometer (BD Biosciences). Data analyses were performed using FlowJo software (Tree Star, Ashland, OR) by gating on live lymphocytes followed by lack of 4′,6-diamidino-2-phenylindole uptake. All additional pregates are indicated and were based on fluorescence minus one controls where applicable.
Statistical analysis
Data were checked for normality using the Shapiro-Wilk normality test. To determine statistical significance between naive and SR1 conditions, a 2-tailed unpaired Student t test was performed using R software. Nonparametric Friedman test with post hoc Dunn’s analysis was performed for cell counts from in vitro expansions. Two-way analysis of variance with post hoc Tukey comparisons was used to determine significant differences within multiple groups. Error bars indicate statistical significance between groups (or between naive and SR1) and are plotted as mean ± standard error of the mean (as indicated in the figure legends).
Results
SR1 can reliably expand progenitors that exhibit T-lymphopoietic potential
We previously reported that naive UCB can generate four- to fivefold expansion of T-cell progenitors.5,6 However, in a transplantation setting requiring a minimum of 2 × 105 CD34+ HSPCs per kilogram of recipient body weight,28,,-31 and with only 4% of UCB units containing a sufficient number of CD34+ cells,32 there is a need to scale up current approaches. Therefore, we investigated whether addition of SR1 (0.75 μM), which leads to the expansion of CD34+ HSPCs,21,22 could provide a larger starting product for in vitro generation of proT cells from HSPCs. We first optimized and characterized the reproducibility of CD34+ HSPC cultures, as previously described (Figure 1A).21 Addition of SR1 significantly enhanced the expansion of total nuclear cells in our cultures by day 15 (Figure 1B; range, 252- to 746-fold), as well as cells enriched for long-term reconstituting potential,21,33 including CD34+CD38−/lo(Figure 1C; range, 157- to 558-fold) and CD34+CD90+ (Figure 1D; range, 170- to 415-fold). Thus, SR1 culture promotes the retention of cells with long-term reconstituting phenotype, consistent with previous findings.21

Analysis of human HSPCs expanded using SR1 in vitro. (A) 5.0 × 103 MACS-enriched CD34+ HSPCs per mL were cultured in the presence of 0.75 μM of SR1 for 15 days before T-progenitor expansion for 14 days on irradiated OP9-DL1 cells. (B) Total nuclear cells for day 7 and day 15 of SR1 culture. (C) Phenotypic analysis by flow cytometry of CD34 and CD38, and corresponding total numbers of CD34+CD38−/lo cells at day 7 or day 15 of SR1 culture. (D) Phenotypic analysis by flow cytometry of CD34 and CD90, and corresponding total numbers of CD34+CD90+ cells at day 7 or day 15 of SR1 culture. Error bars correspond to standard error of the mean. *P < .05, representing statistical significance compared with day 0 as determined by nonparametric Friedman test with post hoc Dunn’s analysis. SFEM, Serum-Free Expansion Medium; TPO, thrombopoietin.
Analysis of human HSPCs expanded using SR1 in vitro. (A) 5.0 × 103 MACS-enriched CD34+ HSPCs per mL were cultured in the presence of 0.75 μM of SR1 for 15 days before T-progenitor expansion for 14 days on irradiated OP9-DL1 cells. (B) Total nuclear cells for day 7 and day 15 of SR1 culture. (C) Phenotypic analysis by flow cytometry of CD34 and CD38, and corresponding total numbers of CD34+CD38−/lo cells at day 7 or day 15 of SR1 culture. (D) Phenotypic analysis by flow cytometry of CD34 and CD90, and corresponding total numbers of CD34+CD90+ cells at day 7 or day 15 of SR1 culture. Error bars correspond to standard error of the mean. *P < .05, representing statistical significance compared with day 0 as determined by nonparametric Friedman test with post hoc Dunn’s analysis. SFEM, Serum-Free Expansion Medium; TPO, thrombopoietin.
SR1 has a known ability to expand the number of colony forming units in culture, suggesting that it promotes the expansion of early multipotent progenitors with erythromyeloid potential.21 Improvement of platelet generation from megakaryocytes,34 natural killer cell production and function,35 and plasmacytoid and myeloid dendritic cell production has also been observed.36 Although immunodeficient mice reconstituted with SR1-expanded HSPCs showed peripheral T- and B-cell reconstitution,21 kinetics and in vitro analyses of lymphoid lineages were limited. Therefore, we sought to assess whether SR1-expanded HSPCs were capable of generating proT cells in vitro using the OP9-DL1 cell coculture system.23 We first compared the input populations between SR1-expanded HSPCs and naive HSPCs (day-0 coculture; Figure 2A). Although CD34 expression was variable (range, 35.8% to 53.8%) after SR1 HSPC expansion, these cells lacked expression of markers of early T-cell differentiation, including CD7, CD5, and CD1a.2,5,37 Analysis of day-14 cocultures revealed that CD7 expression was apparent on CD34+ HSPCs undergoing early T-cell differentiation, with subsequent acquisition of CD5 and CD1a (Figure 2B), defining commitment to the T lineage.5,38 However, we noted a difference regarding the frequency of CD34+ cells, which was higher in day-14 naive HSPC than in SR1 HSPC cocultures, perhaps because of the initial heterogeneity of CD34 expression within the SR1 HSPC input population. Consequently, SR1 HSPC cocultures had significantly fewer CD34+CD7+ cells at day 14 (Figure 2C). SR1 HSPC and naive HSPC cocultures were otherwise comparable, with similar proportions of CD34−CD7+ and CD7+CD5+ cells. CD1a was detected and present on 8% to 10% of the CD7+ cells for both naive HSPC and SR1 HSPC cocultures and corresponded to cells that had downregulated CD34 expression (Figure 2B). Similar results were achieved with OP9-DL4 cocultures (supplemental Figure 1), consistent with previous reports.39 We further observed ∼fourfold expansion of proT cells from SR1 HSPCs by day 14 of coculture, which was not significantly different from the expansion seen in naive HSPC cocultures (Figure 2D). Comparing the combined effects of SR1 HSPC expansion and the cellularity increase during coculture with OP9-DL1 cells, we were able to generate ∼2 × 103 cells from a single starting CD34+ HSPC. Thus, we achieved an overall 200-fold greater expansion in the production of SR1 HSPC–derived proT cells over that of naive HSPCs, taking into account the CD34 content of the UCB unit used for these studies (Figure 2E).
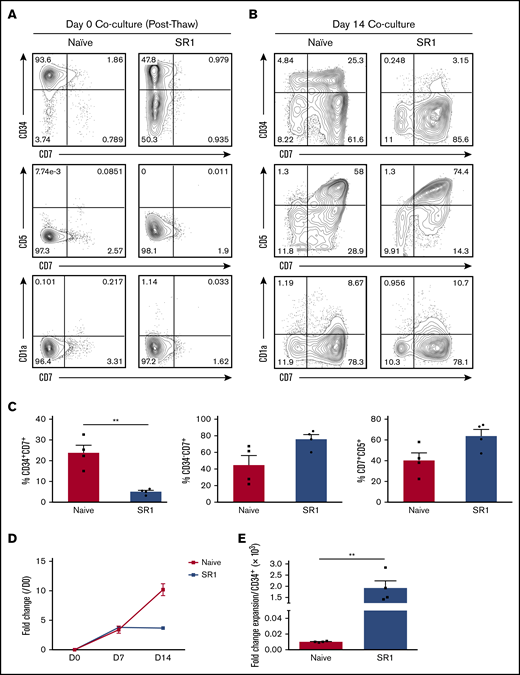
SR1-expanded HSPCs can develop into T-lineage progenitors in vitro. (A) Flow cytometric analysis of CD34, CD7, CD5, and CD1a as markers for early T-progenitor expansion on thawed naive HSPCs or SR1-expanded HSPCs (day-0 coculture with OP9-DL1 cells). (B) Flow cytometric analysis of day-14 coculture for CD34, CD7, CD5, and CD1a expression. The results shown are representative of 4 independent experiments. (C) Percentage of CD34+CD7+, CD34−CD7+, and CD7+CD5+ subsets in naive UCB cocultures compared with SR1 UCB cocultures, shown as a proportion of total live cells in culture. (D) Fold cell expansion of naive vs SR1-expanded HSPC–derived proT cells in OP9-DL1 coculture. (E) Fold cell expansion of naive vs SR1-expanded HSPC–derived proT cells in OP9-DL1 coculture normalized to initial CD34+ input cell number during HSPC expansion (n = 4). Error bars correspond to standard error of the mean. **P < .005, representing statistical significance as determined by unpaired Student t tests.
SR1-expanded HSPCs can develop into T-lineage progenitors in vitro. (A) Flow cytometric analysis of CD34, CD7, CD5, and CD1a as markers for early T-progenitor expansion on thawed naive HSPCs or SR1-expanded HSPCs (day-0 coculture with OP9-DL1 cells). (B) Flow cytometric analysis of day-14 coculture for CD34, CD7, CD5, and CD1a expression. The results shown are representative of 4 independent experiments. (C) Percentage of CD34+CD7+, CD34−CD7+, and CD7+CD5+ subsets in naive UCB cocultures compared with SR1 UCB cocultures, shown as a proportion of total live cells in culture. (D) Fold cell expansion of naive vs SR1-expanded HSPC–derived proT cells in OP9-DL1 coculture. (E) Fold cell expansion of naive vs SR1-expanded HSPC–derived proT cells in OP9-DL1 coculture normalized to initial CD34+ input cell number during HSPC expansion (n = 4). Error bars correspond to standard error of the mean. **P < .005, representing statistical significance as determined by unpaired Student t tests.
SR1 HSPCs generate a unique subset of thymus-homing proT cells in coculture
Thymus-homing cells, identified as CD34+CD7+, have been shown to be present in UCB or fetal bone marrow40 and can be generated in vitro using the OP9-DL1 cell coculture system.5,6 Given our finding that the CD34−CD7+ predominated over the CD34+CD7+ population in SR1 HSPC cocultures, and the fact that the identity of the thymus-seeding progenitor warrants further characterization,2 we tested both of these populations for thymus-reconstituting ability (Figure 3A; gating strategy and purity assessment for isolation of these cells are shown in supplemental Figure 2). As shown in Figure 3B, NSG mice receiving CD34+CD7+ cells generated from either naive or SR1 HSPCs displayed a high frequency of human CD45+ cells in the thymus (95%). Remarkably, we clearly observed detectable engraftment with SR1 CD34−CD7+ cells, whereas CD34−CD7+ cells generated from naive HSPCs failed to engraft. The engraftment with SR1 CD34−CD7+ cells was ∼19-fold lower than its CD34+ counterpart, but nevertheless represents a novel functional capacity of SR1 proT cells, as compared with CD34−CD7+ naive proT cells. Examination of the cell surface phenotype on CD45+ cells revealed that the human cells in mice injected with either subset of cells had progressed along the T-lineage pathway, with CD4+CD8+ double-positive (DP) cells comprising a majority of the human thymocytes present in the engrafted mice (Figure 3B). Thymus cellularity from NSG mice receiving naive HSPC or SR1 HSPC–derived CD34+CD7+ cells or CD34−CD7+ cells is shown in Figure 3C.
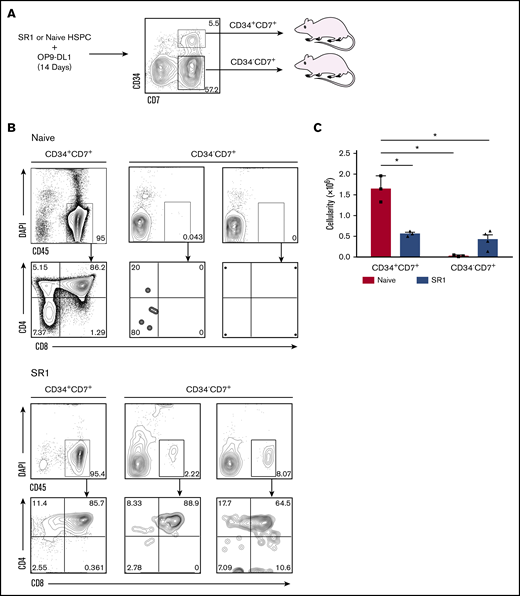
SR1 HSPC–derived CD34+ and CD34− cells that express CD7 engraft neonatal immunodeficient mice. (A) SR1-expanded HSPCs or naive HSPCs were differentiated on OP9-DL1 for 14 days, and CD34+CD7+ and CD34−CD7+ cells were sorted by flow cytometry. Neonatal NSG mice were injected intrahepatically with 1.0 × 106 cells of either subset (n = 3-4 mice per group). (B) Thymuses were harvested after 4 weeks, and cells were flow cytometrically analyzed for CD45, CD4, and CD8 expression. The percentage of live CD45+ cells and CD4 and CD8 are shown for mice transplanted with either subset. (C) Thymus cellularity for transplanted mice. *P < .05, representing statistical significance as determined by 2-way analysis of variance. DAPI, 4′,6-diamidino-2-phenylindole.
SR1 HSPC–derived CD34+ and CD34− cells that express CD7 engraft neonatal immunodeficient mice. (A) SR1-expanded HSPCs or naive HSPCs were differentiated on OP9-DL1 for 14 days, and CD34+CD7+ and CD34−CD7+ cells were sorted by flow cytometry. Neonatal NSG mice were injected intrahepatically with 1.0 × 106 cells of either subset (n = 3-4 mice per group). (B) Thymuses were harvested after 4 weeks, and cells were flow cytometrically analyzed for CD45, CD4, and CD8 expression. The percentage of live CD45+ cells and CD4 and CD8 are shown for mice transplanted with either subset. (C) Thymus cellularity for transplanted mice. *P < .05, representing statistical significance as determined by 2-way analysis of variance. DAPI, 4′,6-diamidino-2-phenylindole.
CD7+ SR1 proT cells home to the thymus in vivo
Our finding that both CD34+CD7+ and CD34−CD7+ SR1 HSPCs have thymus-reconstituting ability led us to redefine the T-cell progenitor generated in SR1 HSPC/OP9-DL1 cell cocultures simply as SR1 CD7+. As anticipated, injection of sorted SR1 CD7+ cells from day-14 cocultures gave rise to high levels of CD45+ thymic engraftment 4 weeks postinjection, with a large number of immature single-positive (SP) and DP cells observed (Figure 4A).
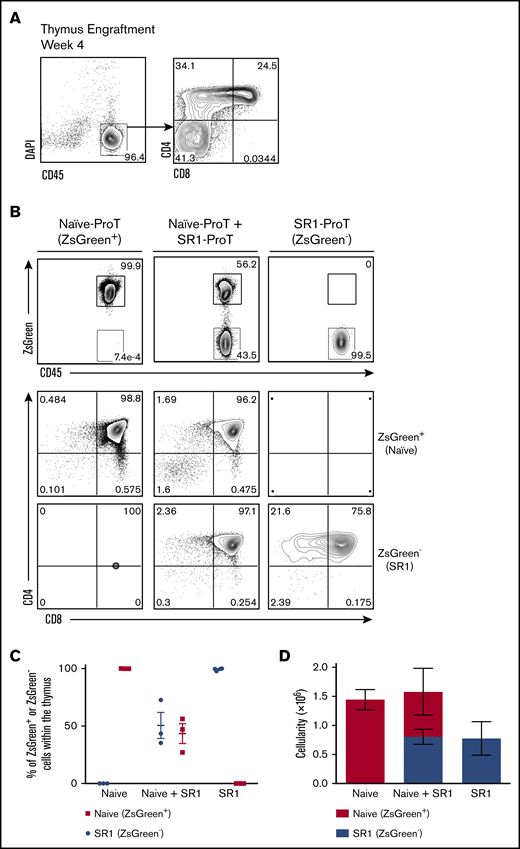
SR1 CD7+ cells home to the thymus in vivo and have comparable homing capabilities to naive CD7+ cells. (A) Thymuses of SR1 CD7+ cell–injected mice were harvested after 4 weeks, and the percentage of live CD45+ cells and CD4 and CD8 are shown (n = 4). (B) A 1:1 mixture of flow cytometrically sorted ZsGreen+ naive CD7+ cells (3.0 × 105) and ZsGreen− (3.0 × 105) SR1 CD7+ cells were injected into nonirradiated NSG neonatal mice, and the thymuses were harvested and analyzed after 4 weeks (n = 3 mice for naive alone or naive plus SR1; n = 4 mice for SR1 alone). Flow cytometric analysis of human CD45 and ZsGreen cell surface expression on DAPI-negative (DAPI−) CD45+–gated cells. CD4 and CD8 expression are shown on CD45+ZsGreen+- and CD45+ZsGreen−-gated cells for naive CD7+ or SR1 CD7+–derived cells, respectively. (C) Percentage of ZsGreen− or ZsGreen+ cells as a proportion of total human CD45+ cells within the thymus for individual mice is shown. (D) Thymic cellularity for ZsGreen− or ZsGreen+ cells in transplanted mice, with error bars corresponding to standard error of the mean.
SR1 CD7+ cells home to the thymus in vivo and have comparable homing capabilities to naive CD7+ cells. (A) Thymuses of SR1 CD7+ cell–injected mice were harvested after 4 weeks, and the percentage of live CD45+ cells and CD4 and CD8 are shown (n = 4). (B) A 1:1 mixture of flow cytometrically sorted ZsGreen+ naive CD7+ cells (3.0 × 105) and ZsGreen− (3.0 × 105) SR1 CD7+ cells were injected into nonirradiated NSG neonatal mice, and the thymuses were harvested and analyzed after 4 weeks (n = 3 mice for naive alone or naive plus SR1; n = 4 mice for SR1 alone). Flow cytometric analysis of human CD45 and ZsGreen cell surface expression on DAPI-negative (DAPI−) CD45+–gated cells. CD4 and CD8 expression are shown on CD45+ZsGreen+- and CD45+ZsGreen−-gated cells for naive CD7+ or SR1 CD7+–derived cells, respectively. (C) Percentage of ZsGreen− or ZsGreen+ cells as a proportion of total human CD45+ cells within the thymus for individual mice is shown. (D) Thymic cellularity for ZsGreen− or ZsGreen+ cells in transplanted mice, with error bars corresponding to standard error of the mean.
To more clearly determine whether SR1 CD7+ cells exhibited comparable thymus-homing capacity to naive CD7+ cells, both subsets were competitively transferred into NSG mice, with the cell of origin traced based on differences in the expression of ZsGreen. In vitro–generated ZsGreen+ naive CD7+ and ZsGreen− SR1 CD7+ cells were injected in a 1:1 ratio (3 × 105 from each subset) into neonatal mice and analyzed 4 weeks postinjection. Figure 4B shows the presence of naive CD7+–derived and SR1 CD7+–derived cells in the thymus of competitively reconstituted mice, as indicated by CD45+ ZsGreen+ (56.2%) and CD45+ ZsGreen− (43.5%) cells, respectively. When examined further, both naive and SR1 CD7+ cells had progressed to the DP stage in competitively reconstituted mice. More importantly, when the percentages of naive proT and SR1 proT cells were analyzed in thymuses across mice, SR1 proT cells were consistently present at comparable frequencies to naive proT cells (Figure 4C). Thymus cellularity from competitively reconstituted NSG mice is shown in Figure 4D.
SR1 CD7+ proT cells differentiate into functionally mature T cells in vivo
Although SR1 proT cells demonstrated strong thymic reconstitution ability, their ability to produce mature, polyclonal human T cells remained to be explored. Upon analyzing NSG mice 10 to 12 weeks postengraftment with SR1 CD7+ cells, we detected recirculating CD4+ and CD8+ SP T lymphocytes within the thymus (Figure 5A), as well as mature circulating CD45+CD3+ T cells in the spleen (Figure 5B). To confirm whether these phenotypically mature peripheral T cells were responsive to stimulation, CD3+ T cells were isolated from the spleens of mice 10 to 12 weeks postengraftment and stimulated with phorbol 12-myristate 13-acetate and ionomycin (Figure 5C) or anti-CD3/CD28 in vitro (Figure 5D). Expression of immunomodulatory cytokines, IL-2, interferon-γ, and TNF-α was observed after stimulation.
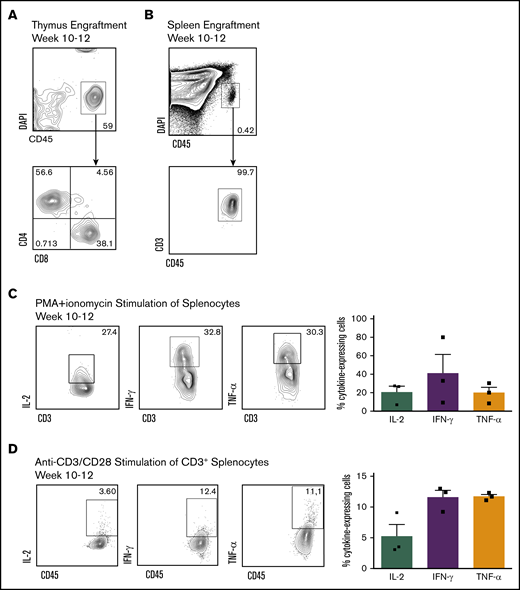
SR1 CD7+ cells are capable of peripheral reconstitution in vivo. (A) Thymuses were harvested 10 to 12 weeks postinjection of SR1 CD7+ cells, and the percentage of live CD45+ cells and CD4 and CD8 are shown (n = 4). (B) Representative flow cytometric plots of CD3 expression on circulating human CD45+ cells harvested from the spleen of mice 10 to 12 weeks after injection of CD7+ cells (n = 6). Representative flow cytometric plots for intracellular IL-2, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) upon in vitro stimulation. Human CD45+CD3+ cells were harvested from the spleen of mice 10 to 12 weeks postinjection of SR1 CD7+ cells, and stimulated for 6 hours with phorbol 12-myristate 13-acetate (PMA)/ionomycin (C) or 72 hours with CD3/CD28 (D). Percentage of cytokine-expressing cells is shown as a proportion of total human CD45+ cells within the thymus for individual mice. Results are from 3 independent experiments, with error bars corresponding to standard error of the mean.
SR1 CD7+ cells are capable of peripheral reconstitution in vivo. (A) Thymuses were harvested 10 to 12 weeks postinjection of SR1 CD7+ cells, and the percentage of live CD45+ cells and CD4 and CD8 are shown (n = 4). (B) Representative flow cytometric plots of CD3 expression on circulating human CD45+ cells harvested from the spleen of mice 10 to 12 weeks after injection of CD7+ cells (n = 6). Representative flow cytometric plots for intracellular IL-2, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) upon in vitro stimulation. Human CD45+CD3+ cells were harvested from the spleen of mice 10 to 12 weeks postinjection of SR1 CD7+ cells, and stimulated for 6 hours with phorbol 12-myristate 13-acetate (PMA)/ionomycin (C) or 72 hours with CD3/CD28 (D). Percentage of cytokine-expressing cells is shown as a proportion of total human CD45+ cells within the thymus for individual mice. Results are from 3 independent experiments, with error bars corresponding to standard error of the mean.
To investigate the molecular nature of these T cells, single-cell RNA sequencing analyses were performed. After filtering out low-quality cells, 1267 naive and 646 SR1 CD3+ cells were analyzed. t-distributed stochastic neighbor embedding analysis of combined cells from both conditions showed 7 unique clusters, labeled C1-7 (Figure 6A). Differential expression analysis was performed to investigate genes that were enriched in each cluster (supplemental Table 2). To further address the intrinsic T-cell heterogeneity, we identified 2 CD8 clusters (C2 and C7) and 5 CD4 clusters (C1, C3, C4, C5, and C6; Figure 6B). The expression of signature genes and known functional markers suggested clusters of CD8+ (naive and effector) and CD4 (naive, effector, and exhausted) cells, and a few clusters were not well defined (Figure 6A). Among the CD4-expressing clusters, C1 was characterized by high expression of exhaustion markers LAG3, TIGIT, and PDCD1, with some cells additionally expressing CTLA-4 (Figure 6C).41 Thus, C1 likely represents exhausted CD4+ T cells. C3 was not readily identifiable but contained cells expressing genes associated with cytotoxicity, including GZMA, GZMK, and IFNG,42 while showing lower expression of T-cell exhaustion markers, such as PDCD1.41 It is likely that this cluster represents heterogeneous T-cell subtypes, including CD4+ effector T cells. Notably, C4 was characterized by high expression of CCR7, IL7R, NELL2, and CD7,43,-45 which are known genes associated with naive T-cell subsets. Clusters 5 and 6 revealed a considerable degree of heterogeneity and therefore were not explicitly defined.
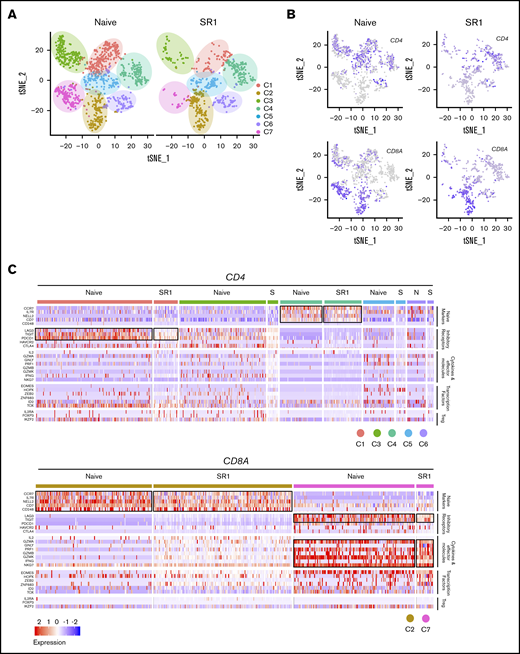
SR1 proT cells can differentiate into heterogeneous T-cell types. (A) t-distributed stochastic neighbor embedding (tSNE) plot showing 7 cell clusters from splenic CD3+ T cells in naive CD7+ or SR1 CD7+ cell–engrafted mice. (B) Analysis of CD4 and CD8A gene expression among CD3+ T cells derived from both naive CD7+ or SR1 CD7+ cell–engrafted mice. (C) Heatmap showing expression of T-cell function–associated genes in each cluster. Results are filtered based on CD4 or CD8A expression, as indicated. Black boxes highlight the prominent patterns defining known T-cell subtypes (n = 3 mice pooled for each group). Treg, regulatory T cell.
SR1 proT cells can differentiate into heterogeneous T-cell types. (A) t-distributed stochastic neighbor embedding (tSNE) plot showing 7 cell clusters from splenic CD3+ T cells in naive CD7+ or SR1 CD7+ cell–engrafted mice. (B) Analysis of CD4 and CD8A gene expression among CD3+ T cells derived from both naive CD7+ or SR1 CD7+ cell–engrafted mice. (C) Heatmap showing expression of T-cell function–associated genes in each cluster. Results are filtered based on CD4 or CD8A expression, as indicated. Black boxes highlight the prominent patterns defining known T-cell subtypes (n = 3 mice pooled for each group). Treg, regulatory T cell.
We next looked at the CD8A-expressing clusters. C7 was defined as CD8+ effector T cells, because it was characterized by high expression of chemokines CXCL13 and CCL5, and genes associated with cytotoxicity, including high expression of PRF1, GZMA, and GZMK (Figure 6C; supplemental Table 2). In contrast, C2 lacked expression of effector genes and predominantly expressed genes associated with naive T cells, including CCR7, IL7R, NELL2, CD7, and CD248.43,,-46 These data suggest considerable functional diversity in CD4+ and CD8+ T-cell subsets in both naive and SR1 proT-cell recipients.
SR1 CD7+ proT cells develop into polyclonal T cells
A major concern after HSCT is an inability to completely restore a polyclonal T-cell repertoire, which may never reach pretransplantation levels.47 Therefore, we wanted to understand whether the functional peripheral T cells generated from SR1 proT cells were clonally diverse. CD3+ T cells isolated from the periphery of engrafted mice were analyzed by single-cell RNA sequencing. Clonotypes with ambiguous gene alignments were filtered out before assessing their V-(D)-J chain usages, with different combinations of rearrangements denoting a unique T-cell clonotype. As shown in Figure 7A, the number of effective clonotypes that were represented in naive CD7+–injected mice (510 clones) was similar to that in SR1 CD7+ mice (531 clones), as determined by the True Diversity Index.
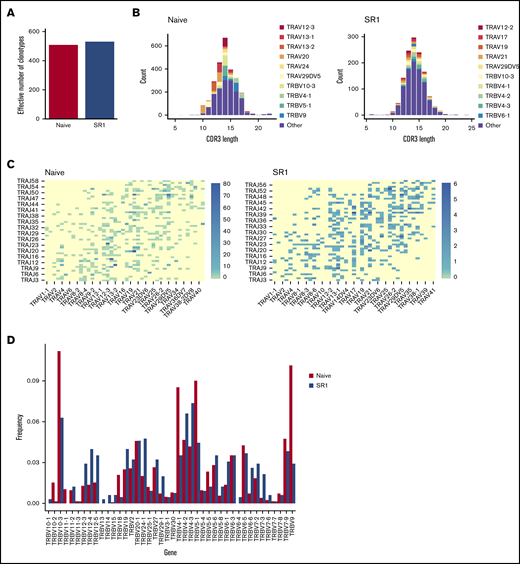
SR1 CD7+ cells generate a polyclonal T-cell repertoire. (A) Effective number of TCR clonotypes found in the spleen of naive CD7+ or SR1 CD7+ cell–engrafted mice, as determined by the True Diversity Index. Results are normalized to account for differences in the number of sequenced cells between conditions. (B) CDR3 spectratype for spleen-derived CD3+ cells from mice injected with naive proT cells or SR1 proT cells. The distribution of TCRα genes per CDR3 length is shown. (C) Heatmap representation of percentage of TRA V to J (oriented via chromosomal 5′ to 3′ distribution) pairings among total sequences in naive (left) and SR1 (right) T-lineage cells derived from the periphery of mice injected with either naive CD7+ cells or SR1 CD7+ cells. (D) TCRβ gene diversity in splenocytes from immunodeficient mice transplanted with day-14 naive or SR1 CD7+ cells.
SR1 CD7+ cells generate a polyclonal T-cell repertoire. (A) Effective number of TCR clonotypes found in the spleen of naive CD7+ or SR1 CD7+ cell–engrafted mice, as determined by the True Diversity Index. Results are normalized to account for differences in the number of sequenced cells between conditions. (B) CDR3 spectratype for spleen-derived CD3+ cells from mice injected with naive proT cells or SR1 proT cells. The distribution of TCRα genes per CDR3 length is shown. (C) Heatmap representation of percentage of TRA V to J (oriented via chromosomal 5′ to 3′ distribution) pairings among total sequences in naive (left) and SR1 (right) T-lineage cells derived from the periphery of mice injected with either naive CD7+ cells or SR1 CD7+ cells. (D) TCRβ gene diversity in splenocytes from immunodeficient mice transplanted with day-14 naive or SR1 CD7+ cells.
Greater precision of diversity estimation was achieved via spectratyping analysis,48,,-51 where the number of different CDR3 lengths is used as a proxy for the number of clonotypes. Variable normal distribution of CDR3 lengths was observed for both the naive and SR1 conditions, confirming diversity in both the naive CD7+– and SR1 CD7+–derived repertoires (Figure 7B). The SR1 CD7+–derived repertoire used different TCRα genes compared with the naive CD7+–derived repertoire, whereas both encoded variable CDR3 amino acid lengths (Figure 7B). Analysis of TCRα rearrangements showed a similar usage of distal and proximal Vα to Jα gene elements (Figure 7C). Examination of Vβ families showed that some Vβ chains were represented in only the naive condition (TRBV11-1) and others only in the SR1 condition (TRBV12-5, TRBV13, and TRBV6-3; Figure 7D). Nevertheless, there was broad representation of diverse Vβ chains, further confirming that mature CD3+ TCRαβ T cells displaying a polyclonal TCR repertoire were present in the periphery of both naive and SR1 CD7+ recipients after transplantation.
Discussion
Because T-cell lymphopenia is a critical risk factor for patient infections and relapse post-HSCT, managing T-cell reconstitution using an allogeneically compatible transplantation strategy remains important. HSPC expansion using SR1 allowed us to increase the amount of HSPCs from a single UCB unit, enabling the generation of proT cells from the same unit. ProT cells derived from SR1-expanded HSPCs showed similar thymic engraftment compared with their naive counterparts, while retaining an ability to generate a multifunctional and polyclonal peripheral T-cell repertoire in vivo. Our findings, combined with the known ability of SR1-expanded HSPCs to generate erythromyeloid lineages,21 including platelets,34 natural killer cells,35 and plasmacytoid and myeloid dendritic cells,36 have important implications for conferring rapid multicellular immunity post-HSCT.
An important consideration for a therapeutic proT-cell product is its immunophenotypic definition. To this end, we and others have traditionally defined proT cells as CD34+CD7+ cells,5,6,40,52 as corroborated by the ability of this population to home to the thymus within immunodeficient hosts. However, compared with naive HSPCs, SR1 HSPCs developed into predominantly CD34−CD7+ cells after 14 days of coculture with OP9-DL1 cells. The novel finding that SR1 HSPC–derived CD7-expressing CD34+ and CD34− populations both engrafted the thymus in vivo allowed us to immunophenotypically redefine proT cells simply as CD7+CD4−CD8−CD3−, which constitutes a larger proT-cell product that can be used for adoptive transfer in the clinic. The capacity of 2 distinct subsets to home to the thymus is interesting in light of a recent study performed using mouse T-cell progenitors, where it was shown that different mouse proT-cell subsets had distinct roles in enhancing thymic function.53 Indeed, our previous findings also highlight that proT cells have a role beyond thymus seeding, because they are capable of inducing changes within the thymic microenvironment,6 thus prompting the investigation of the roles for CD34+CD7+ vs CD34−CD7+ subsets.
Although both naive and SR1 proT cells engrafted the thymus of recipients, we noted differences in thymic cellularity and phenotypic outcomes. SR1 proT cells engrafted the thymus at levels that resulted in overall lower thymic cellularity, although this is likely explained by the difference in the temporal kinetics, as evidenced by the presence of CD4 immature SP cells and a lower proportion of DP cells at the time points analyzed. To more clearly determine whether 1 subset or another exhibited better thymic engraftment capacity, we placed SR1 proT cells in direct competition with naive proT cells in vivo. Our present findings indicate that SR1 proT cells displayed comparable thymic engraftment when placed in competition with naive proT cells, despite the lower thymic cellularity achieved by injecting SR1 proT cells alone (Figure 4D). A possible explanation for these findings is that the SR1 proT cells have differential thymic homing capacities compared with naive proT cells. In keeping with this notion, we observed that although SR1 HSPC–derived CD34−CD7+ cells were able to engraft, they did so at a slightly reduced capacity compared with CD34+CD7+ cells. Given that the SR1 CD7+–refined proT-cell product contained mostly CD34−CD7+ cells, this difference in engraftment is perhaps not surprising.
Homing capacity may be a function of cellular abilities to respond to thymic chemokine gradients and adhesion molecules through cell surface expression of CCR7, CCR9, and PSGL-1,54,,-57 which may serve to provide a potential mechanism for the decreased cellularity seen in recipients of SR1 proT cells. Alternatively, SR1 proT cells may have a proliferative or survival disadvantage compared with naive proT cells when injected alone because of impairments in crosstalk activity with thymic epithelial cells.58 These interactions are predominantly mediated by TNF superfamily receptor/ligand interactions. Our previous work6 has shown that naive proT cells express the TNF superfamily ligand RANK ligand at high levels, perhaps providing a means to support thymic crosstalk for SR1 proT cells when both cell types are coinjected during competitive reconstitution.59,60 The absolute ability to generate >200-fold more proT cells using a single SR1-expanded HSPC compared with a naive HSPC would enable us to overcome the apparent thymic engraftment and/or cellularity differences afforded by the SR1 proT cells by simply injecting more cells.
The effective appearance of SR1 proT cells at the level of the thymus led us to investigate whether these cells could generate a peripheral T-cell repertoire that was both functionally diverse and polyclonal. An ongoing concern with HSPC expansion strategies is a loss of HSC clonality or expansion of facilitator, nonstem HSPC compartments, which have the potential to affect downstream immune lineage outcomes. True HSC clonality has been difficult to assess because of the limited suitable in vivo transplantation assays for testing functionality of a single human HSC.21 Our findings suggest that irrespective of HSC clonality, T-cell clonality is unaffected given that intrathymic development and selection of proT cells are contingent on only a small number of HSC clones for the generation of a polyclonal T-cell repertoire.61 Therefore, immune competence after HSCT is not related to the absolute number of HSCs present, but rather to intrathymic events. In keeping with this notion, we observed a polyclonal T-cell repertoire by CDR3 spectratyping and Vα/Vβ chain usage analysis in the periphery of mice injected with either naive or SR1 proT cells. However, it is important to note that this allowed us to describe the TCR repertoire only at the time of evaluation within a homeostatic, nonchallenged immune state. The number of clonotypes are often a drastic underestimate of TCR diversity, which can on average be 10 to 100× greater.48,51,62 Furthermore, only major clones (>0.1%) were likely detected in samples with active TCR transcription.63
Importantly, our single-cell RNA sequencing revealed functional heterogeneity among cells within the polyclonal CD3+ T-cell compartment. We focused on both CD4- and CD8A-expressing CD3+ T-cell clusters. Compared with mice that received naive proT cells, mice injected with SR1 proT cells revealed comparable expression of genes defining CD4-expressing exhausted T cells (C1),41 as well as CD4- or CD8A-expressing cytotoxic T cells (C3 and C7)42 and naive T cells (C2 and C4). An important limitation of our study was an inability to recapitulate the full spectrum of T-cell subset heterogeneity, including memory T-cell responses. For formation of these cell subsets, antigenic challenge and cellular support from other cell types of the immune system, including myeloid and B cells, are required. Therefore, directly testing our cell types under antigenic challenge in an SR1 HSCT model constitutes an important next step.
Because SR1 HSPCs have already been clinically tested,22 a detailed implementation of SR1 HSPC–derived proT cells in patients undergoing HSCT warrants further study. In summary, we provide evidence that SR1 HSPCs combined with SR1 HSPC–derived proT cells seems to be a promising therapeutic option for overcoming lymphopenia for patients undergoing HSCT. SR1 HSPC expansion may also be a useful strategy for amplifying genetically modified HSCs in a tumor immunotherapy approach.64,65
Acknowledgments
The authors thank Génève Awong and Courtney McIntosh for their expertise in cell sorting, Lisa Wells and Christina R. Lee for their technical support with animal experiments, and Elaine Herer and Rose Kung from the Women and Babies Program at Sunnybrook Health Sciences Centre (Toronto, ON, Canada) for their ongoing support by providing umbilical cord blood.
This work was supported by grants from the Canadian Institutes of Health Research (FND154332), the Ontario Institute for Regenerative Medicine, the Krembil Foundation, Medicine by Design: a Canada First Research Excellence Fund Program at the University of Toronto, and an Innovation to Impact grant from the Canadian Cancer Society (#705960) (J.C.Z.-P.); from the National Cancer Institute, National Institutes of Health (2P01 CA142106) (B.R.B.); from the Children’s Cancer Research Fund (H.E.S. and B.R.B.); and from the National Institute of Child Health and Human Development, National Institutes of Health (K12-HD068322), and St. Baldrick’s Foundation (H.E.S.).
Authorship
Contribution: J.S. designed and performed the experiments, analyzed all data, and wrote the manuscript; E.L.Y.C. performed in vitro experiments, analyzed the single-cell RNA sequencing data, and discussed the results; Y.X. performed in vitro experiments; H.E.S. designed and performed some in vitro and in vivo experiments and edited the manuscript; and B.R.B. and J.C.Z.-P. provided critical experimental advice and edited the manuscript.
Conflict-of-interest disclosure: The authors have submitted a patent describing the method of producing and using SR1 proT cells. H.E.S. receives remuneration as a consultant and advisor to the Novartis Speaker Bureau, and as a consultant for Jazz Consulting. B.R.B. receives remuneration as an advisor to Kamon Pharmaceuticals, Inc., Five Prime Therapeutics, Inc., Regeneron Pharmaceuticals, Magenta Therapeutics, and BlueRock Therapeutics; research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences, Inc., AbbVie, Inc., BlueRock Therapeutics, Leukemia and Lymphoma Society, Childrens' Cancer Research Fund, and KidsFirst Fund; and is a cofounder of Tmunity. J.C.Z.-P. is an advisor to Intellia Therapeutics and is a cofounder of Notch Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Juan Carlos Zúñiga-Pflücker, Sunnybrook Research Institute, 2075 Bayview Ave, Room M7-619, Toronto, ON M4N 3M5, Canada; e-mail: jczp@sri.utoronto.ca; or Bruce R. Blazar, Division of Blood and Marrow Transplantation, Department of Pediatrics, University of Minnesota, MMC 366 Mayo, 8366A (Campus Delivery Code), 420 Delaware St SE, Minneapolis, MN 55455; e-mail: blaza001@umn.edu.
The full-text version of this article contains a data supplement.

