

Key Points
Upon injury, the mesothelium recruits neutrophils to the peritoneal space, which contributes to adhesion formation.
Neutrophil recruitment and macrophage-depletion kinetics in adhesions differ from the normal innate response.

Abstract
Peritoneal adhesions are pathological fibroses that ensnare organs after abdominal surgery. This dense connective tissue can cause small bowel obstruction, female infertility, and chronic abdominal pain. The pathogenesis of adhesions is a fibrotic response to tissue damage coordinated between mesothelial cells, fibroblasts, and immune cells. We have previously demonstrated that peritoneal adhesions are a consequence of mechanical injury to the mesothelial layer sustained during surgery. Neutrophils are among the first leukocytes involved in the early response to tissue damage. Here, we show that when subjected to mechanical stress, activated mesothelial cells directly recruit neutrophils and monocytes through upregulation of chemokines such as CXCL1 and monocyte chemoattractant protein 1 (MCP-1). We find that neutrophils within the adhesion sites undergo cell death and form neutrophil extracellular traps (NETosis) that contribute to pathogenesis. Conversely, tissue-resident macrophages were profoundly depleted throughout the disease time course. We show that this is distinct from traditional inflammatory kinetics such as after sham surgery or chemically induced peritonitis, and suggest that adhesions result from a primary difference in inflammatory kinetics. We find that transient depletion of circulating neutrophils significantly decreases adhesion burden, and further recruitment of monocytes with thioglycolate or MCP-1 also improves outcomes. Our findings suggest that the combination of neutrophil depletion and monocyte recruitment is sufficient to prevent adhesion formation, thus providing insight for potential clinical interventions.
Introduction
Adhesions are bands of fibrous tissue that anchor organs to each other or the peritoneal wall and are a common cause of significant postoperative morbidity. Clinical sequelae include small bowel obstruction, chronic pain, female infertility, and even death.1,,,,,,-8 Most studies focus on the later stages of adhesion formation, involving the initiation of the clotting cascade and fibrin deposition to reinforce the final adhesion. However, studies dealing with the early stages are often less characterized, although evidence implicates interactions between the mesothelium, fibroblasts, and hematogenous cells.9,-11
We recently demonstrated that insult to the surface mesothelium initiates adhesion formation. Abrasion of the peritoneal wall leads to the proliferation of the mesothelial layer through a hypoxia-inducible factor 1α program, upregulation of fetal surface markers such as mesothelin, and an expansive change in gene expression. Ultimately, these changes cause the outward growth of the mesothelium and its derived fibroblasts into the peritoneal cavity, resulting in an adhesion.12
Although mesothelial cells and their progeny play a key role, it is likely that other cell types contribute to adhesion formation. It has been shown that leukocytes infiltrate early into the peritoneal injury site and play a role in the inflammatory cascade that includes classical proinflammatory signals such as transforming growth factor β and interferon γ.13,14 We therefore hypothesized that modulation of the inflammatory process is a potential avenue for adhesion prevention. Previous studies have shown that a broadly neutralizing antineutrophil serum or IFNγ blockade can attenuate adhesion formation.10,11,13 However, the mechanism of recruitment, the cell types and cellular dynamics involved, and the function of blood cells in adhesion sites have not been fully characterized. A better understanding of these processes is necessary to discover novel targets and develop more efficacious therapies.
Models of sterile inflammation, such as thioglycolate administration, have been shown to cause acute responses from leukocytes.15 After intraperitoneal infusion of thioglycolate, neutrophils are rapidly recruited to the peritoneal cavity, reaching their peak numbers 4 hours after infusion.16 Then, neutrophils begin to apoptose and their levels decline significantly by 12 hours postinfusion, becoming negligible by 24 hours. Monocyte/macrophage levels decrease during the first few hours after infusion, but then increase by 4 hours and reach their peak after 2 days. In this model, macrophages execute programmed cell removal (PrCR), phagocytosing apoptotic neutrophils around the time neutrophils begin intracellular DNA breakdown, to prevent tissue damage from the cellular debris.15 Whether adhesion formation reproduces these same kinetics and mechanisms has yet to be shown.
We have previously shown that a subset of mesothelial cells differentiate in a progenitor-like fashion.12 In pathological conditions, these mesothelial cells drive progression and severity of adhesion by directly contributing to fibrosis in the days following injury.17 Here, we show that the mesothelium also serves as an immune modulator in the first few hours after injury and recruits inflammatory blood cells in the initial stages of adhesion formation. We further demonstrate that neutrophils and resident macrophages play opposite roles in the pathogenesis of adhesions. Specifically, neutrophils, which were thought to cause damage through granule release and production of free-radicals,11 directly contribute to adhesion formation through apoptosis and the subsequent formation of neutrophil extracellular traps (NETs). Macrophages play an opposite, protective role, likely by the PrCR of apoptotic and necrotic neutrophils. Lastly, we show that targeted depletion of circulating neutrophils, disruption of NET formation, and recruitment of monocytes leads to better outcomes in a mouse model of surgical adhesions.
Materials and methods
Adhesion induction and scoring
Adhesion-induction surgeries were performed on wild-type C57BL/6J (The Jackson Laboratory) mice aged 6 to 10 weeks and scored as previously described in Tsai et al.12
Parabiosis
Parabiosis surgeries were done on age-matched (4- to 6-week-old) female wild-type C57BL/6J (The Jackson Laboratory) and C57BL/Ka Rosa26 mRFP1 mice. Mice to undergo parabiosis were housed together in a single cage for 14 days prior to surgery. Mice were anesthetized by inhaled isoflurane until determined unconscious as confirmed by the toe-pinch test. The sides of the mice were shaved and cleaned with 70% ethanol and betadine. Mice were laid next to each other and incisions from elbow to knee were made on adjacent sides. The elbow and knee joints were ligated using 4-0 sutures (Ethicon) and the loose skin from adjacent mice was stapled together. Mice were allowed to recover on a heating pad and injected daily with 0.05 to 0.1 mg/kg buprenorphine. Mice were followed closely and monitored daily for signs of morbidity for 14 days. Staples were removed following 14 days and retro-orbital blood was assayed at that time for chimerism.
Histology and immunofluorescence
Immunofluorescence studies were performed as previously described in Tsai et al.12
Peritoneal lavages
Adhesion-induction surgeries were done on wild-type C57BL/6J (The Jackson Laboratory) mice aged 6 to 10 weeks. Following induction, mice were euthanized and subsequently given a 5-mL intraperitoneal injection of lavage solution (phosphate-buffered saline [PBS] with 2% fetal bovine serum and 2 mM EDTA) and counting beads, along with 3 mL of air. The intraperitoneal fluid was agitated to capture cells and then 1.5 mL of the solution was extracted with an 18G syringe, while being careful to avoid inducing hemorrhage. The peritoneal lavage was analyzed by flow cytometry to measure absolute count of various immune cell types.
Luminex assay
Peritoneal lavage was performed as described in the previous section with a lavage solution comprising PBS with 2 mM EDTA and protease inhibitors. Peritoneal fluid (1.5 mL) was harvested from the cavity as previously described in "Peritoneal lavages" and centrifuged gently at 1200 rpm for 5 minutes. The supernatant was removed and underwent a second centrifugation at 2000g, from which 0.5 mL was taken for analysis.
Flow cytometry
Peritoneal washes were filtered through a 100-µm filter, spun at 1200 rpm for 5 minutes, and washed with 2% fetal bovine serum with 2 mM EDTA in PBS. Cells were stained with anti-Gr1 (peridinin-chlorophyll protein complex Cy5.5), anti-CD45 (fluorescein isothiocyanate), anti-CD11b (BV605), anti-Ter119 (allophycocyanin), and anti-F4/80 (phycoerythrin) and analyzed.
Antibody treatments
Adhesions were induced in wild-type C57BL/6J (The Jackson Laboratory) and allowed to recover for 7 days. Monoclonal anti–Gr-1 (1A8) (200 µg; BioXCell) antibody was administered via retro-orbital injections 24 hours prior to surgery. Recombinant monocyte chemoattractant protein-1 (MCP-1) (1 µg; R&D Biosystems) was added via intraperitoneal injections immediately following surgery. Mice were euthanized 7 days after initial surgery and scored for adhesion severity.
Peripheral blood and tissue harvest and cell extraction
Blood was extracted from treated and control mice via retro-orbital bleed and analyzed in a HemaTrue (Heska) analyzer for blood cell counts. Citrate buffer (1×, with salts) was used to perfuse mice, and peripheral blood was collected in a syringe until the liver became pale. Peripheral blood was centrifuged, aspirated, vortexed to resuspension in residual fluid, and lysed in ACK lysis buffer for 4 minutes. Upon completion of the reaction, the supernatant was aspirated and cells were resuspended in the remaining fluid, of which 1 mL was taken for flow cytometry. The liver and lung were ground and incubated for 30 minutes in 1 mL of a 2× solution of Liberase TL and DNAse in Hank's balanced salt solution (HBSS)–containing calcium and magnesium. Bone marrow from femurs was extracted and filtered through a 100-µm filter with HBSS containing calcium and magnesium. The spleen was similarly pushed through a filter with a syringe plunger and salt-containing HBSS. All tissue samples were centrifuged at 1200 rpm for 5 minutes. Blood supernatant was aspirated, and cells were resuspended in residual liquid. Liver, lung, bone marrow, and spleen were lysed in 1.5 mL of ACK lysis buffer for 1 minute, quenched with PBS, and centrifuged again. All samples were then resuspended in fluorescence-activated cell sorter buffer (spleen in 400 µL; the bone marrow, liver, and lung in 200 µL each), of which 75 µL were taken for staining with anti-Gr1 (fluorescein isothiocyanate), anti-CD11b (phycoerythrin Texas Red), and anti-CD45 (allophycocyanin). Following a second wash after 25 minutes of staining, cells were resuspended in 200 µL of fluorescence-activated cell sorter buffer containing 4′,6-diamidino-2-phenylindole and taken for flow cytometry.
Experimental approval
All animal experiments were carried out in strict accordance with the guidelines set forth by the Association for Assessment and Accreditation of Laboratory Animal Care International, Stanford University’s Administrative Panel on Laboratory Animal Care (protocol number 10266), and the United States.
Results
We previously developed a mouse model for surgical adhesions and used it to map the transcriptional changes of the surface mesothelium in the immediate and early periods (up to 24 hours) after injury in adhesion formation.12 Briefly, peritoneal cavities are exposed and an ischemic button is placed and lightly abraded with a surgical brush, while leaving the mesothelial layer intact. At the same time, sham controls, which involve a peritoneal laparotomy, are done without button placement or abrasion, providing an important distinction between a generalized injury response and an adhesion-inducing injury response.
We hypothesized that soluble mediators may play a role in coordinating the immune response after mechanical injury. Using RNA sequencing, we found that the injured mesothelium upregulates a battery of proinflammatory cytokines and chemokines, whose expression peaks within 6 hours (Figure 1A; supplemental Figure 1). Specifically, the mesothelium strongly upregulates CXCL1 (GROA) (50-fold; P = .0012), CCL2, or MCP-1 (CCL2) (690-fold; P = .0006), and CXCL2 (1660-fold; P = .0086) early after adhesion induction. Although these cytokines peaked after 6 hours, they remained significantly upregulated after 24 hours (supplemental Figure 1).
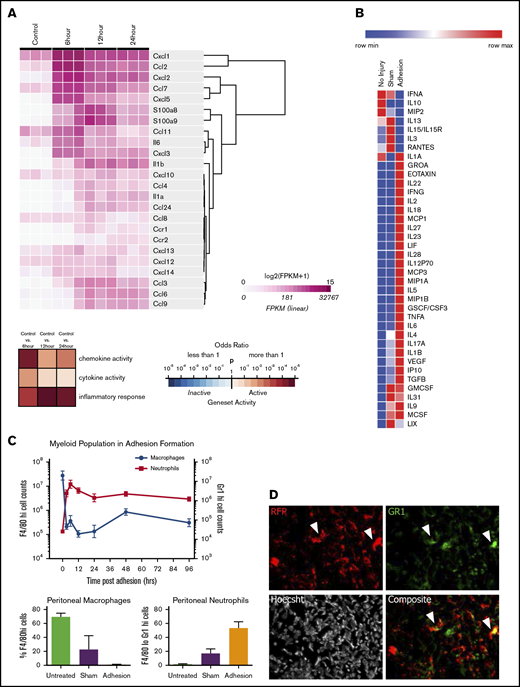
The mesothelium recruits neutrophils and macrophages through during adhesion induction. (A) Heatmap of immune active proteins from RNA sequencing of purified surface mesothelium immediately after and 6, 12, and 24 hours after button placement and clustered by gene expression. (B) Heatmap of Luminex multiplex assays of key immune active proteins. (C) Characterization of myeloid population from 0 to 96 hours following adhesion formation (top). Absolute numbers of macrophages (blue) and neutrophils (red) were analyzed at each time point. (Bottom) Percentage of F4/80hi and Gr-1hi cells after 24 hours in untreated, sham surgery, or adhesion-induced mice. (D) Single and composite immunofluorescence staining for Gr-1 of adhesions from wild-type mice of a wild-type/RFP+ parabionts pair. Immunofluorescence stain. Magnification ×20. Arrows denote RFP+Gr-1+ cells. CSF, colony-stimulating factor; FPKM, fragments per kilobase of transcript per million mapped reads; GMCSF, granulocyte macrophage colony-stimulating factor; GSCF, granulocyte colony-stimulating factor; IFN, interferon; IL, interleukin; IP, interferon γ–induced protein; LIF, leukemia inhibitory factor; LIX, CXCL5; max, maximum; MCSF, macrophage colony-stimulating factor; min, minimum; MIP, macrophage inflammatory protein; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
The mesothelium recruits neutrophils and macrophages through during adhesion induction. (A) Heatmap of immune active proteins from RNA sequencing of purified surface mesothelium immediately after and 6, 12, and 24 hours after button placement and clustered by gene expression. (B) Heatmap of Luminex multiplex assays of key immune active proteins. (C) Characterization of myeloid population from 0 to 96 hours following adhesion formation (top). Absolute numbers of macrophages (blue) and neutrophils (red) were analyzed at each time point. (Bottom) Percentage of F4/80hi and Gr-1hi cells after 24 hours in untreated, sham surgery, or adhesion-induced mice. (D) Single and composite immunofluorescence staining for Gr-1 of adhesions from wild-type mice of a wild-type/RFP+ parabionts pair. Immunofluorescence stain. Magnification ×20. Arrows denote RFP+Gr-1+ cells. CSF, colony-stimulating factor; FPKM, fragments per kilobase of transcript per million mapped reads; GMCSF, granulocyte macrophage colony-stimulating factor; GSCF, granulocyte colony-stimulating factor; IFN, interferon; IL, interleukin; IP, interferon γ–induced protein; LIF, leukemia inhibitory factor; LIX, CXCL5; max, maximum; MCSF, macrophage colony-stimulating factor; min, minimum; MIP, macrophage inflammatory protein; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
To confirm that this pattern of gene expression corresponds to protein secretion, a multiplex protein assay was done on peritoneal lavages 1, 3, and 7 days following adhesion induction. Adhesions were induced as previously described in "Adhesion induction and scoring" and mice were euthanized at 1, 3, and 7 days after surgery. Luminex multiplex assays were done on peritoneal washes, which demonstrated increased levels of CXCL1 (threefold increase over uninjured controls; P = .004) and MCP-1 (12-fold increase over controls; P = .0205) (Figure 1B; supplemental Figure 2) as well as other chemoattractants such as eotaxin (61-fold increase over control; P = .031), macrophage inflammatory protein 1B (MIP1B) (18-fold increase over sham; P = .0343), interleukin 18 (IL-18) (100-fold increase over sham; P = .0001), and IL-6 (380-fold increase over control; P = .0272). Interestingly, CXCL2 protein levels were decreased (3.43-fold decrease over control; P = .0071). These factors were also changed compared with sham surgeries, suggesting that adhesion formation elicits a very specific inflammatory cascade.
Finally, to demonstrate blood cell recruitment, peritoneal infiltrates were immunophenotyped by flow cytometry. Washes from mice undergoing adhesion induction were collected and analyzed for Gr-1, CD11b, and F4/80 expression18 (Figure 1C). Neutrophils (Gr-1+ cells) rapidly infiltrated into the peritoneal cavity within 6 hours and remained present for over 96 hours after adhesion formation. Neutrophil concentrations in the peritoneal cavity were found to be 3 times those of sham controls after 24 hours. Inflammatory monocytes (Cd11b+F4/80mid) increased quickly in concentration following damage, never returning to their initial levels (supplemental Figure 3). Tissue-resident macrophages (CD11b+F4/80hi cells), however, follow an inverse trend, rapidly decreasing in number within 6 hours and slowly increasing, although never reaching their preoperative concentration. The decrease in macrophage concentration in adhesions was 25 times less than in sham controls after 24 hours.
To compare this to traditional acute inflammatory models, we parabiosed CFP+ and RFP+ mice and allowed them to achieve chimerism for 14 days. Thioglycolate was injected into the CFP+ peritoneal cavity, and peritoneal and circulating blood cells from both mice were analyzed after 5 hours or 3 days (supplemental Figures 4-6). We observed heavy neutrophil infiltration into the peritoneum that had received the injection after 5 hours, which resolved after 3 days (supplemental Figure 4). Infiltrating neutrophils were in part RFP+, indicating their circulatory origin. At the same time, monocytes infiltrate and stay at increased levels until 3 days (supplemental Figure 4). Monocytes were overenriched in the peritoneum over the blood, hinting at either a tissue source, preferential recruitment from the peritoneum, and or a difference in time of residence or survival in peritoneum than blood (supplemental Figure 6). Tissue-resident macrophages did not decrease after thioglycolate infusion, but instead increased (supplemental Figure 4).
The pattern of inflammatory cell recruitment in peritoneal adhesion formation differs from that of thioglycolate as previously found and discussed in the above paragraph. In thioglycolate-induced sterile peritonitis, macrophages begin as the predominant cell type in the peritoneum. Several hours after injection of thioglycolate, neutrophils are recruited to the peritoneal cavity, exceeding macrophages in abundance before gradually decreasing in number until they once again become undetectable 72 hours after inflammation.18 In adhesions, neutrophils persist in the peritoneal cavity 96 hours after their initial entry and are neither eliminated from the site nor adequately replaced by macrophages after the resolution of the inflammation. Although both thioglycolate-induced peritonitis and adhesions are manifestations of peritoneal inflammation, the kinetics of immune cell recruitment are markedly different, likely due to the degree of damage leading to the sustained removal of peritoneal macrophages and the ongoing recruitment of neutrophils.
Neutrophils are well known for playing a proinflammatory role, which results in not only microbial protection but also tissue repair. Granulopoiesis occurs in the bone marrow, and mature neutrophils exit into the blood stream (circulating pool) and subsequently take up temporary residence in solid tissues (marginated pool). Based on the magnitude of increased neutrophil counts, we hypothesized that a number of neutrophils observed in the peritoneal cavity after injury are migrants from the circulation rather than tissue-resident sources. To confirm that some cells at adhesion sites were derived from the circulation, parabiosis experiments were performed between reporter-labeled (RFP+) and wild-type mice, and blood chimerism was confirmed after 14 days (supplemental Figure 7A). Adhesions were induced in the wild-type parabionts, and tissue was harvested and adhesions analyzed 7 days later. Adhesions contained high levels of RFP+ cells (Figure 1D; supplemental Figure 7B), confirming that circulating blood cells (macrophages or neutrophils) infiltrate into the adhesion site. Note that Gr-1+ and F4/80+RFP− and Gr-1+RFP+ cells were also found in adhesion sites (Figure 1D; supplemental Figure 7B), consistent with the chimerism in the blood.
To assess whether adhesion severity could be modulated, mice undergoing adhesion induction were pretreated with known conditions that alter the recruitment of inflammatory cells. As mentioned previously, thioglycolate has been shown to cause peritonitis and elicits an inflammatory response in a highly characteristic manner.15 This results in the effective recruitment of monocytes to the peritoneal cavity 24 hours after thioglycolate administration. We hypothesized that a thioglycolate-induced alteration of the immune environment (depletion of circulating neutrophils and an increase in effective concentration of peritoneal macrophages) prior to adhesion induction could lead to a decrease in adhesion formation.
Mice were injected with thioglycolate 3 days prior to adhesion induction surgery. Adhesions were assessed 7 days after surgery and scored on a scale from 0 to 6 (0 corresponding to no adhesions and 6 corresponding to death caused by adhesions) as previously described in Tsai et al.12 Mice receiving thioglycolate (n = 17) 3 days prior to injury were found to have a significantly decreased adhesion burden compared with controls (supplemental Figure 8). Mice receiving thioglycolate prior to surgery have more peritoneal macrophages at the time of potential adhesion initiation. Monocyte recruitment may result in a greater capacity for PrCR to clear activated neutrophils recruited to the peritoneum at the time of surgery, and ultimately decrease the fibrotic response. This suggests that modulation of the immune system is a potentially effective method for preventing adhesions.
To better understand the contributions of neutrophils in isolation in adhesion formation, we first treated mice the day prior to surgery with an IV injection of a depleting anti–Gr-1 antibody (clone 1A8). Whereas the Gr-1 clone RB6-8C5 also binds to monocytes and macrophages, 1A8 has been shown to be a more specific antibody to target neutrophils and granulocytes19 and 1A8 has been shown to transiently deplete circulating neutrophils. To understand the kinetics of neutrophil depletion, we injected 200 µg of antibody IV and then measured neutrophil levels over the subsequent hours and days (Figure 2A) in the blood and other tissues. Neutrophil depletion from the blood occurs steadily in the first 24 hours postinfusion, but numbers start to recover by 48 hours postinfusion, likely reflecting reflex hematopoiesis. As previously stated, neutrophils exist in multiple compartments, such as in the bone marrow, spleen, and the interstitial space of other solid tissues. To understand which tissue compartments were affected by 1A8 treatment, we analyzed the percentage of neutrophils in spleen, bone marrow, liver, and lung tissue. Interestingly, we found that at our injected dose of antibody, only the circulating neutrophils were depleted, leaving the percentage of splenic, bone marrow, and hepatic neutrophils largely unaffected (Figure 2A; supplemental Figure 9).

NETs play a role in adhesion induction, and depletion of NETs and neutrophils result in adhesion prevention. (A) Percentage of neutrophils in peripheral blood 6, 12, 24, and 48 hours following anti–Gr-1 administration. (B) Adhesion scoring of adhesion-induced mice treated with PBS (n = 63), Gr-1 alone (n = 4), MCP-1 alone (n = 5), and Gr-1 combined with MCP-1 (n = 14). (C) Single and composite immunofluorescence staining for H3Cit and Hoescht in adhesion tissue 7 days following adhesion induction. White arrows denote areas highly concentrated for nuclear stain and H3Cit suggestive of NETs. (D) Single and composite immunofluorescence staining for Gr-1 and H3Cit in wild-type/RFP+ parabiotic mice. (C-D) Immunofluorescence stain. Scale bar, 100 µm. (E) Adhesion scoring of adhesion-induced mice treated with intraperitoneal injections of PBS (n = 4) or DNase I (n = 5) or osmotic pumps filled with PBS (n = 5) or DNase I (n = 5). NS, not significant.
NETs play a role in adhesion induction, and depletion of NETs and neutrophils result in adhesion prevention. (A) Percentage of neutrophils in peripheral blood 6, 12, 24, and 48 hours following anti–Gr-1 administration. (B) Adhesion scoring of adhesion-induced mice treated with PBS (n = 63), Gr-1 alone (n = 4), MCP-1 alone (n = 5), and Gr-1 combined with MCP-1 (n = 14). (C) Single and composite immunofluorescence staining for H3Cit and Hoescht in adhesion tissue 7 days following adhesion induction. White arrows denote areas highly concentrated for nuclear stain and H3Cit suggestive of NETs. (D) Single and composite immunofluorescence staining for Gr-1 and H3Cit in wild-type/RFP+ parabiotic mice. (C-D) Immunofluorescence stain. Scale bar, 100 µm. (E) Adhesion scoring of adhesion-induced mice treated with intraperitoneal injections of PBS (n = 4) or DNase I (n = 5) or osmotic pumps filled with PBS (n = 5) or DNase I (n = 5). NS, not significant.
Given that 1A8 depletes only circulating granulocytes and shows effects most prominently at 24 hours postinjection, we injected animals IV 24 hours before surgery and assessed adhesion formation 7 days later. Mice receiving 1A8 anti–Gr-1 (n = 5) were found to develop significantly less severe adhesions than mice receiving PBS injections (adhesion index ∼1 vs 3-4, respectively). Our results indicate that depletion of circulating neutrophils is sufficient to significantly decrease adhesion burden. Therefore, it is likely that this pool of inflammatory neutrophils is a necessary agent in adhesion formation.
Given the lower adhesion severity after thioglycolate injection, we sought to test whether inflammatory monocytes contributed to the resolution of mechanical injury. To test monocytes in isolation of other cell types, we injected 1 µg of recombinant MCP-1 intraperitoneally, which specifically recruits monocytes into the peritoneal cavity, where they can differentiate predominantly into macrophages.20 When MCP-1 was administered immediately following surgery, we found a reduction in adhesion severity, with MCP-1–injected mice scoring around 1 to 2 compared with 3 to 4 in PBS-treated controls (Figure 2B).
To test the synergy between these cell-mediated effects, mice receiving a combination treatment of anti–Gr-1 and MCP-1 were found to have significantly fewer and milder adhesions (n = 14) than vehicle controls or than either single agent in isolation. (Figure 2B). This indicates that neutrophils and macrophages play a key role in adhesion formation. Depletion of neutrophils or recruitment of monocytes leads to a moderate reduction of adhesion burden, but the combined effects of neutrophil depletion and monocyte recruitment leads to a more significant outcome.
To understand the mechanism by which neutrophils contribute to adhesion formation, sections of adhered tissues were analyzed by microscopy. We first observed that Gr-1+ neutrophils in adhesion sites differed in morphology (more elongated) from traditional neutrophils (small, circular cells) (Figure 1D), suggesting that circulating neutrophils are not only simply migrating into adhesion sites, but also likely undergoing some change.
Interestingly, dense, highly cellular areas were often observed within adhesions, in between organs and peritoneum (Figure 2C; supplemental Figure 10), and spaced out along tissue borders. These areas were highly Hoechst-positive, indicating high amounts of DNA. Analysis with anticitrullinated histone 3 (H3Cit) showed highly specific staining in these dense regions, which is an indicator of NETs.21,22 Furthermore, these areas stained positively for Gr-1 and associated with Gr-1+ cells (Figure 2D; supplemental Figure 7). After inducing adhesions in the nonlabeled parabiont of a pair of RFP+ and wild-type mice, we observed that RFP+ circulating Gr-1+ cells can undergo NETosis after recruitment by mesothelium (Figure 2D).
To determine whether disruption of NETs could affect adhesion stability, mice receiving adhesion induction were treated with DNase I either through intraperitoneal injection (50 µg every 12 hours for 7 days) or through a continuous release format using an osmotic pump placed in the peritoneal cavity that releases ∼20 µg of DNase I every hour for 7 days (Figure 2E). Adhesions were assessed 7 days after surgery and scored according to a modified version of our previously described scheme (see “Methods”). DNase I administered through constant release of an osmotic pump resulted in a significant decrease in adhesion burden (Figure 2E). Although it was found that the presence of the osmotic pump alone exacerbates adhesion formation, from a score of 3.75 ± 0.5 to 6.0 ± 0.0 with a PBS pump, a DNase I pump reduced adhesion severity to 5.0 ± 0.5 from 6.0, and NETs were not visualized on adhesion sections by microscopy. These data suggest that although NETosis is a contributing mechanism, disruption of NETs with DNase is not sufficient to eliminate adhesions.
To further understand the role of NETs in adhesion formation, we obtained PADI4-deficient mice, which lack peptidyl arginine deiminase 4 (PADI4), the necessary enzyme for conversion of arginine residues on histone 3 to citrulline, a process that decondenses chromatin and allows for the formation of NETs. We induced adhesions in PADI4−/− and in wild-type mice and examined the tissue after 7 days. Similar to our previous finding, PADI4−/− mice had significantly milder adhesions, although this background was not sufficient enough to completely abolish adhesions (supplemental Figure 11).
These findings suggest that the injured mesothelium initiates and sustains recruitment of neutrophils from the circulation for days after the initial injury, which exacerbate peritoneal adhesions through inflammatory mechanisms that include but are not limited to NETosis. After mechanical injury to the adhesion site, macrophages are sustainably depleted from the peritoneal cavity. By modifying both responses, we observe a highly significant reduction in adhesion burden with potential clinical implications.
Discussion
The pathogenesis of adhesions has been extensively studied but has only recently been characterized cellularly and molecularly through RNA sequencing and lineage tracing. We have previously shown that the mesothelium, through a stem/progenitor-like mechanism, is a cell of origin that initiates adhesion formation.12 However, it is clear that the mesothelium is not the only cell type involved. Previous studies have detailed the proinflammatory contributions of blood lineage cells that exacerbate adhesions. Here, we show that the mesothelium also plays an immune-regulatory role in which it recruits circulating monocytes and granulocytes, and activates peritoneal macrophages.
When compared with benign models of inflammation such as thioglycolate, peritoneal adhesions have a prolonged inflammatory course. During thioglycolate-induced inflammation, neutrophils infiltrate rapidly and return to baseline levels within 24 hours, while peritoneal macrophages are depleted and replenished in a similar amount of time. In our adhesion model, neutrophils infiltrate from the circulation and their numbers remain higher in the peritoneal space for much longer than in thioglycolate-induced peritonitis. Our data show that the mesothelium, after extensive injury, continues to secrete cytokines and chemokines long after the injury, prolonging the recruitment of neutrophils.
At the same time, our data suggest that peritoneal macrophages are initially depleted in adhesion formation, but never return to their normal levels. This prolonged depletion has many potential causes, including the mechanics of surgery, difficulty with intravasation of either or both blood cell types, issues in the engraftment of these cells after intravasation, or the limited regenerative capacity of local resident macrophages derived from yolk sac progenitors.23,24 The absence of macrophages could diminish the rate and efficacy of PrCR, leading to the accumulation of neutrophilic apoptotic bodies and NETs and contributing to adhesion formation. Therefore, it follows that the adhesion-formation process is partially a result of differences in neutrophil/monocyte kinetics compared with other models of inflammation, and may occur in response to neutrophil frequency and overactivity. We find that inflammatory monocytes follow similar kinetics as neutrophils, but their effect on adhesion formation has yet to be understood. Our data show that the observed monocyte population does not significantly contribute to the tissue-resident macrophage pool and therefore appears to play a distinct role in the fibrotic process.
NETs have traditionally been associated with microbial defense25,26 ; it is thought that the adhesiveness of free DNA leads to entrapment of pathogens. High concentrations of DNA have been known to increase the viscosity of bronchoalveolar fluid in cystic fibrosis patients, for whom administration of DNAse I has been shown to reduce viscosity and improve clinical outcomes.27,28 More recently, studies have shown the increasing importance of NETs in other disease processes such as cancer and diabetes.29,-31 Our findings show the presence of NETs at the interface between 2 tissues in an adhesion (Figure 2C-D) in which treatment with DNase I significantly reduces severity and mortality, likely suggesting that neutrophils use DNA in the form of NETs to increase adhesiveness between tissue surfaces. Our studies with PADI4−/− mice similarly showed benefit, suggesting that NETs are likely 1 aspect of adhesion pathogenesis. This builds upon our previous findings in which we identify the mesothelium as the key regulator and cellular component that produces adhesions. Our current findings hint at a mechanism in which the mesothelium is not only a cell of origin, but also an immune regulator able to recruit neutrophils that activate and strengthen the final adhesion.
The evidence that modulation of neutrophil and macrophage peritoneal concentrations leads to adhesion prevention indicates that circulating blood cells play a critical role. We have shown that neutrophils take part in the adhesion-forming process through their infiltration and NETosis. The sustained absence of macrophages likely hinders any PrCR that usually occurs in inflammation, further contributing to the resultant adhesion. Correction of the adhesion-related neutrophil-dominant inflammatory imbalance of cell levels through preoperative treatment with anti–Gr-1 and MCP-1 is able to prevent adhesions. We predict that these inflammatory cells may also interface and/or signal with the mesothelium before the irrevocable outward expansion of the mesothelial cell progeny deposits in adhesive fibroses. The identity of these signals will be important to further elucidate, as they likely represent a local soluble target for antiadhesion therapies.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank R. Nusse, J. Sage, P. Beachy, P. Berg, C. Wang, A. McCarty, G. Wernig, and J. Chen for helpful discussions.
This work was supported in part by the Virginia and D. K. Ludwig Fund for Cancer Research, the National Heart, Lung, and Blood Institute of the National Institutes of Health under award numbers R01HL058770 and U01HL099999 (I.L.W.), and grants from the California Institute of Regenerative Medicine (RC1 00354) and the Smith Family Trust (I.L.W.). J.M.T. was supported by the National Institutes of Health, National Institute of General Medical Sciences (T32GM007365), the Ruth L. Kirschstein National Research Service Award (1F30DK108561 [National Institute of Diabetes and Digestive and Kidney Diseases]), and by the Paul and Daisy Soros Fellowship for New Americans. N.B.F. was supported by Ruth L. Kirschstein postdoctoral fellowship F32HL115963-02 (National Heart, Lung, and Blood Institute).
Authorship
Contribution: J.M.T., N.B.F., B.M.G., and M.S. designed experiments with suggestions from I.L.W.; J.M.T., B.M.G., M.S., and N.B.F. performed all experiments and analyses; R.S. performed RNA sequencing and analysis; J.S. provided the bioinformatics analysis platforms; K.D.M., M.N.M., K.S.K., A.K.V., and M.M. performed experiments; Y.R. provided experimental suggestions and suggestions for the manuscript; and J.M.T. and M.S. wrote the manuscript with suggestions from N.B.F. and I.L.W.
Conflict-of-interest disclosure: J.M.T., N.B.F., Y.R., R.S., M.N.M., and I.L.W. have applied for patents on these topics. The remaining authors declare no competing financial interests.
Correspondence: Irving L. Weissman, Lokey Stem Cell Institute, Stanford University Medical Center, 275 Campus Dr, Stanford, CA 94305-5324; e-mail: irv@stanford.edu; or Jonathan M. Tsai, Department of Pathology, Brigham and Women’s Hospital, 75 Francis St, Boston, MA 02115; e-mail: jmtsai@bwh.harvard.edu.

