

Key Points
Immune recognition and processing of PGN induces monocyte TF expression.
A positive proinflammatory feedback signaling reinforces the primary monocyte procoagulant responses to PGN.

Abstract
Disseminated intravascular coagulation is a frequent manifestation during bacterial infections and is associated with negative clinical outcomes. Imbalanced expression and activity of intravascular tissue factor (TF) is central to the development of infection-associated coagulopathies. Recently, we showed that anthrax peptidoglycan (PGN) induces disseminated intravascular coagulation in a nonhuman primate model of anthrax sepsis. We hypothesized that immune recognition of PGN by monocytes is critical for procoagulant responses to PGN and investigated whether and how PGN induces TF expression in primary human monocytes. We found that PGN induced monocyte TF expression in a large cohort of healthy volunteers similar to lipopolysaccharide stimulation. Both immune and procoagulant responses to PGN involve intracellular recognition after PGN internalization, as well as surface signaling through immune Fcγ receptors (FcγRs). In line with our hypothesis, blocking immune receptor function, both signaling and FcγR-mediated phagocytosis, significantly reduced but did not abolish PGN-induced monocyte TF expression, indicating that FcγR-independent internalization contributes to intracellular recognition of PGN. Conversely, when intracellular PGN recognition is abolished, TF expression was sensitive to inhibitors of FcγR signaling, indicating that surface engagement of monocyte immune receptors can promote TF expression. The primary procoagulant responses to PGN were further amplified by proinflammatory cytokines through paracrine and autocrine signaling. Despite intersubject variability in the study cohort, dual neutralization of tumor necrosis factor-α and interleukin-1β provided the most robust inhibition of the procoagulant amplification loop and may prove useful for reducing coagulopathies in gram-positive sepsis.
Introduction
Disseminated intravascular coagulation (DIC) is an acquired syndrome commonly associated with sepsis or trauma, and it represents a negative prognostic marker for these pathologies.1 Pathophysiology of DIC involves tissue factor (TF)–dependent activation of blood coagulation, depression of anticoagulant mechanisms, and inhibition of fibrinolysis.2 TF binds and activates plasma coagulation factor VII (FVII), and the TF-FVIIa tenase complex is the main initiator of clotting reactions in hemostasis and thrombosis.3 Expression of TF in the intravascular compartment is low in healthy humans4,5 but elevated in patients with sepsis6 and multiple experimental sepsis models.7-10 Monocyte TF expression is critical for coagulopathies associated with sepsis,11 and intravascular TF neutralization has been explored therapeutically.12-14
Lipopolysaccharide (LPS) is the main procoagulant pathogen-associated molecular pattern (PAMP) in DIC associated with gram-negative sepsis.7,15 Despite similar incidence of DIC in gram-positive sepsis,1 procoagulant PAMPs in gram-positive pathologies are less understood. LPS is scarcely represented in gram-positive pathogens, but Toll-like receptor 2 (TLR2) activation by cell wall lipoteichoic acid,16 peptidoglycan (PGN)-associated lipoproteins,17 or bacterial superantigens18 have been shown to induce procoagulant responses in vitro. Unlike LPS, synergism of 2 gram-positive PAMPs19,20 or with endotoxin21 was required for coagulopathic manifestations in experimental models in vivo.
Thrombosis and/or consumptive coagulopathies have been documented in human22-24 and animal25 cases of anthrax. These pathologies are recapitulated by nonhuman primate experimental models26-28 but less represented in rodents.29 We recently showed that Bacillus anthracis–derived PGN infusion in nonhuman primates mimics the pathophysiology of live B anthracis IV challenges27 and promotes DIC and associated organ damage.30 Similar consumptive coagulopathies have been observed in rats challenged with anthrax PGN but not lethal or edema exotoxins.31 Anthrax PGN is a glycan polymer of repeating disaccharide units of N-acetylglucosamine and N-acetylmuramic acid cross-linked by peptide stem bridges,32 which induces robust proinflammatory responses by innate immune phagocytes.33-36 Immune responses to anthrax PGN are TLR-independent37 and driven by the cytosolic sensors NOD1 and NOD2 (nucleotide-binding oligomerization receptors 1 and 2, respectively)38,39 or the NLRP3 inflammasome.40 As such, opsonization of PGN,36,41,42 recognition by immune receptors,36,41 phagocytosis, and lysosomal processing34,35 are prerequisites for robust immune responses to PGN. Alternatively, immunoglobulin-opsonized PGN can initiate proinflammatory signaling by clustering and activating immunoglobulin Fcγ receptors (FcγRs) at the cell surface.36,41,42 Ultimately, both NOD-dependent43 as well as surface FcγR signaling44 lead to activation of nuclear factor κB B (and/or activator protein 1 transcription factors, which are also the main drivers of TF transcription during inflammation.45
We recently reported that PGN induces DIC in vivo through activation of both arms of the clotting cascade, and we confirmed intravascular expression of TF in PGN-challenged nonhuman primates.30 Here, we investigated the mechanism by which PGN triggers TF expression in primary human peripheral blood mononuclear cells (PBMCs) and found that PGN is a potent procoagulant PAMP. PGN promoted overlapping proinflammatory and procoagulant responses that mimicked monocyte stimulation by the parental pathogen. Both internalization-dependent and surface FcγR signaling were required for robust monocyte TF expression. Unlike LPS stimulation, primary procoagulant responses to PGN were amplified by the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) as cytokine neutralization reduced the amplification of PGN procoagulant responses.
Materials and methods
Analysis of primary human monocytes
Studies on primary human PBMCs were approved by the Institutional Review Board at the Oklahoma Medical Research Foundation. Voluntary participants were informed of study aims and procedures, and provided written informed consent. PBMCs were isolated on the day of the experiment by density gradient centrifugation using Histopaque-1077 (MilliporeSigma, St. Louis, MO). Unless otherwise specified, PBMCs were rested in RPMI 1640 containing 1% autologous human serum for 1 hour at 37°C in a humidified atmosphere containing 5% carbon dioxide, then stimulated for up to 6 hours with either PGN (10 µg/mL, purified as described elsewhere37 ), LPS (1 µg/mL from Escherichia coli serotype O55:B5, TLR4-specific agonist; Enzo Life Sciences, Farmingdale, NY), Pam3CSK4 peptide (1 µg/mL synthetic lipopeptide TLR2 agonist; Enzo Life Sciences), or heat-killed B anthracis (107 CFU/mL). In select experiments, pharmacologic inhibitors were added during the PBMC resting step and kept throughout the experiment.
Flow cytometry analysis of procoagulant and proinflammatory responses
PBMCs were stimulated in temperature-responsive Nunc UpCell 96-well plates (Thermo Fisher Scientific, Waltham, MA). After stimulation, plates were transferred to 4°C for 20 minutes to detach adhered cells and immediately fixed with 4% paraformaldehyde. Cellular determinants and inducible antigens were detected by immunostaining of saponin-permeabilized PBMCs. Monocytes were identified based on size, scatter, and CD14 expression (clone 61D3 immunoreactive). Procoagulant responses were defined by TF antigen expression (clone HTF-1 immunoreactive), and proinflammatory responses were defined by TNF-α expression (clone Mab11 immunoreactive). Paired labeled antibodies and isotype controls (clone P3.6.2.8.1) were from eBioscience (San Diego, CA). Data were collected using by either LSR II or FACSCelesta cytometers (BD Biosciences, San Jose, CA).
Pharmacologic interrogation of signaling networks
Internalization-dependent PGN signaling was blocked by the phagocytic inhibitor cytochalasin D (cytoD; 15 µM; EMD Millipore, Billerica, MA). FcγR-mediated cell surface PGN signaling was blocked with piceatannol (50 µM; Enzo Life Sciences), a Syk kinase inhibitor. Proinflammatory signaling feedback was blocked by using neutralizing monoclonals against TNF-α (infliximab; Janssen Biotech, Horsham, PA), IL-6 receptor (tocilizumab; Genentech, South San Francisco, CA), or IL-1β (clone 4H5; InvivoGen, San Diego, CA).
Supernatant transfer studies
PBMCs were stimulated with PGN or LPS for up to 6 hours as described. At the end of the primary stimulation, cells (donors) were processed for flow cytometry while supernatants (conditioned media) were spun 10 minutes at 21 000g to eliminate residual PGN particles, then stored overnight at 4°C. Naive PBMCs (recipients) were isolated the next day, rested in RPMI 1640 for 1 hour, and then cultured in the presence of supernatants saved from donor cells for 6 hours. TF expression in PGN-stimulated and/or conditioned media–stimulated monocytes was subsequently analyzed by using flow cytometry.
Data analysis and representation
Flow cytometry analysis, gating, and histogram visualization were performed in FlowJo (Tree Star, Ashland, OR). Data analysis was performed by using Prism (GraphPad Software, La Jolla, CA). Normal or log-normal distribution of primary responses to PAMPs and, when appropriate, outlier identification using the ROUT method46 were performed in Prism. Differences between the 2 groups were analyzed by using paired Student t tests. Differences between multiple groups were analyzed by repeated measures (RM) analysis of variance (ANOVA) with either Tukey’s (1-way ANOVA) or Holm-Sidak’s (2-way ANOVA) multiple comparison tests for normally distributed data, or Friedman’s test with Dunn’s multiple comparison test for nonnormal distributed data. The statistical significance threshold was set at P < .05 (*P < .05, **P < .01, ***P < .001, and ****P < .0001; ns = not significant). Unless otherwise specified, individual responses and median and interquartile range (IQR; horizontal bars) are depicted for either the frequency of responsive CD14+ monocytes (either TF+ and/or TNF-α+ monocytes) or antigen expression (individual values represent geometric mean of TF or TNF-α relative fluorescence intensities). Graphs were generated with Prism and figures collated in Adobe Illustrator (Adobe Systems, San Jose, CA).
Results
PGN induces monocyte TF as potently as LPS
We investigated the procoagulant potential of PGN in primary human monocytes using a large cohort of healthy volunteers (n = 50 independent donors; male and female subjects; age range, 20-60 years). For comparison, a saturating concentration of LPS, the established procoagulant PAMP in Gram infections, was used as control. As expected, absent PAMP stimulation, TF expression was minimal in PBMCs isolated from healthy volunteers (median [IQR], 0.78% [0.14%-3.0%]) (Figure 1). TF expression increased after stimulation of primary human PBMCs with either LPS (1 µg/mL) or PGN (10 µg/mL) and was restricted to CD14+ monocytes. TF antigen expression in unstimulated donors did not follow a normal distribution, and a small fraction of donors presented with low basal levels of monocyte TF (17%; 15 of 86 independent measurements) and identified as outliers according to the ROUT method.46 In subsequent assessments (2-4 for each donor, ≥1 month apart), basal TF expression was not detected at least one half of the times measured in every one of these volunteers, with one exception (donor withdrew from the study); this scenario highlights the transient nature of basal TF expression in these donors. In response to PAMP stimulation, we observed significant induction of monocyte TF antigen in all donors studied, including individuals expressing basal monocyte TF. There was no significant difference between the fraction of TF+CD14+ monocytes after either PGN or LPS stimulation (median [IQR], 46.4% [33.4%-60.1%] and 47.6% [33.5%-60.3%], respectively). Likewise, monocyte TF fluorescence intensity was similar between PGN- and LPS-treated cells and approximately fivefold higher than in unstimulated cells (median [IQR] TF fluorescence intensities of 766 [411-1259], 741 [421-1006], and 141 [36-227]). We found a strong correlation between the amplitude of PGN- and LPS-induced TF expression in paired donor samples, indicating similar procoagulant responses to gram-positive (PGN) and gram-negative (LPS) PAMPs.
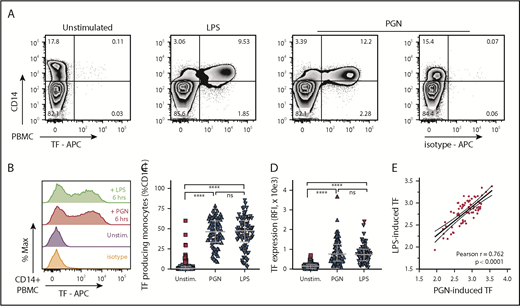
PGN induces TF expression in primary human monocytes in vitro. (A) Exemplification of PBMC procoagulant responses induced by stimulation with either PGN (10 µg/mL) or LPS (1 µg/mL) for 6 hours, as identified by TF expression in CD14+ monocytes in a median responsive individual. (B) Histogram overlay of TF expression in CD14+ PBMCs after PAMP stimulation of a median responsive individual. (C) PAMP induction of procoagulant monocytes (TF+CD14+) in the human cohort studied (n = 86 independent measurements). Horizontal bars depict median and IQR, and outliers are identified by red symbols. (D) Monocyte TF antigen expression (geometric mean of fluorescence intensity) in PAMP-stimulated PBMCs (n = 86). Median, IQR, and outlier representation is consistent with panel C. (E) Correlation between LPS- and PGN-induced procoagulant responses in the study cohort. Individual data represent paired, log-normalized, TF fluorescence intensities of LPS- or PGN-stimulated PBMCs assessed in parallel. Best-fit linear regression (continuous line) and 95% confidence intervals (dotted lines) are graphically represented, and Pearson correlation coefficient (r) is shown in inset. APC, allophycocyanin.
PGN induces TF expression in primary human monocytes in vitro. (A) Exemplification of PBMC procoagulant responses induced by stimulation with either PGN (10 µg/mL) or LPS (1 µg/mL) for 6 hours, as identified by TF expression in CD14+ monocytes in a median responsive individual. (B) Histogram overlay of TF expression in CD14+ PBMCs after PAMP stimulation of a median responsive individual. (C) PAMP induction of procoagulant monocytes (TF+CD14+) in the human cohort studied (n = 86 independent measurements). Horizontal bars depict median and IQR, and outliers are identified by red symbols. (D) Monocyte TF antigen expression (geometric mean of fluorescence intensity) in PAMP-stimulated PBMCs (n = 86). Median, IQR, and outlier representation is consistent with panel C. (E) Correlation between LPS- and PGN-induced procoagulant responses in the study cohort. Individual data represent paired, log-normalized, TF fluorescence intensities of LPS- or PGN-stimulated PBMCs assessed in parallel. Best-fit linear regression (continuous line) and 95% confidence intervals (dotted lines) are graphically represented, and Pearson correlation coefficient (r) is shown in inset. APC, allophycocyanin.
PGN induction of monocyte TF is dose- and time-dependent
PGN induced TF in primary human monocytes in a dose-dependent manner (Figure 2A). We reported that PGN opsonization by serum proteins was necessary for efficient internalization and processing of PGN and supported proinflammatory responses to PGN.36,42 Likewise, PGN induction of TF was enhanced by the presence of human serum. We observed a 30% to 50% reduction of estimated EC50s (50% effective concentrations) at higher serum concentration (10%) compared with low serum (1%). Similarly, PGN induced monocyte TF expression in a time-dependent manner (Figure 2B). After PGN stimulation, TF antigen was continuously induced within the 6-hour time-course of the current study. In contrast, LPS stimulation achieved maximal TF antigen expression by 4 hours (supplemental Figure 1). Furthermore, analysis of donor variability indicated that roughly one-half of the tested donors induced TF as early as 2 hours (fast responders), whereas others showed significant TF expression by 6 hours only (slow responders).

PGN induces monocyte TF expression in a dose- and time-dependent manner. (A) PGN induces monocyte TF expression in a dose-dependent manner. Representative overlaid histograms of a median responsive individual (left) and average (n = 8 independent donors) monocyte procoagulant responses after 6 hours stimulation with PGN (0-50 µg/mL; middle and right panels). Procoagulant responses were measured in the presence of autologous serum at either high (10%; blue squares) or low (1%; red circles) concentration. Best-fit 4-parameter logistic curves used to estimate EC50 are depicted by continuous or dotted lines, respectively. (B) PGN (10 µg/mL) induces monocyte TF expression in a time-dependent manner. Representative overlaid histograms of a median responsive individual (left) and distribution of TF procoagulant responses (middle and right panels) in the cohort studied (n = 14). Individual time-course responses are identified by identical symbols, and horizontal bars indicate median and IQR of procoagulant responses to PGN in the cohort studied. RFI, relative fluorescence intensity.
PGN induces monocyte TF expression in a dose- and time-dependent manner. (A) PGN induces monocyte TF expression in a dose-dependent manner. Representative overlaid histograms of a median responsive individual (left) and average (n = 8 independent donors) monocyte procoagulant responses after 6 hours stimulation with PGN (0-50 µg/mL; middle and right panels). Procoagulant responses were measured in the presence of autologous serum at either high (10%; blue squares) or low (1%; red circles) concentration. Best-fit 4-parameter logistic curves used to estimate EC50 are depicted by continuous or dotted lines, respectively. (B) PGN (10 µg/mL) induces monocyte TF expression in a time-dependent manner. Representative overlaid histograms of a median responsive individual (left) and distribution of TF procoagulant responses (middle and right panels) in the cohort studied (n = 14). Individual time-course responses are identified by identical symbols, and horizontal bars indicate median and IQR of procoagulant responses to PGN in the cohort studied. RFI, relative fluorescence intensity.
PGN induces monocyte TF through surface and internalization-dependent signaling
Proinflammatory responses to PGN require internalization and lysosomal processing, followed by activation of either cytosolic NOD receptors or inflammasome. Immunoglobulin G (IgG)–opsonized PGN can also activate surface FcγRs on immune cells. Blocking PGN internalization using inhibitors of phagocytosis such as cytoD (Figure 3) or latrunculins (not shown) reduced the monocyte procoagulant responses to PGN by 65% to 75% (P = .0078, RM-ANOVA with Dunnett’s posttest). FcγR-mediated events, both signaling and Fcγ-mediated phagocytosis, require signal transduction through Syk tyrosine kinase. Consequently, pharmacologic inhibition proximally downstream of FcγR using piceatannol, a Syk kinase inhibitor, also reduced PGN-dependent TF expression by ∼75% (P = .0007, RM-ANOVA with Dunnett’s posttest). Combined inhibition of phagocytosis and FcγR function effectively abrogated monocyte procoagulant responses to PGN. Similar results were obtained in the presence of either autologous serum or fetal bovine serum supplemented with purified human IgG, without affecting the variability of donor responses. Thus, intersubject variability in the studied cohort relates more to the intrinsic monocyte processing/signaling ability and not to the opsonin titers in the serum of the different donors. Paired analysis of procoagulant (TF) and proinflammatory (TNF-α) outcomes in a subset of our donors indicated that both responses occur through the same signaling avenues. Procoagulant responses to PGN were less sensitive to cytoD than the paired proinflammatory responses (P = .0059, RM-ANOVA with Holm-Sidak’s posttest), indicating that cell surface FcγR signaling (cytoD-insensitive but piceatannol-sensitive) might preferentially promote primary procoagulant responses, whereas internalization-mediated signaling is required for both proinflammatory and procoagulant responses.
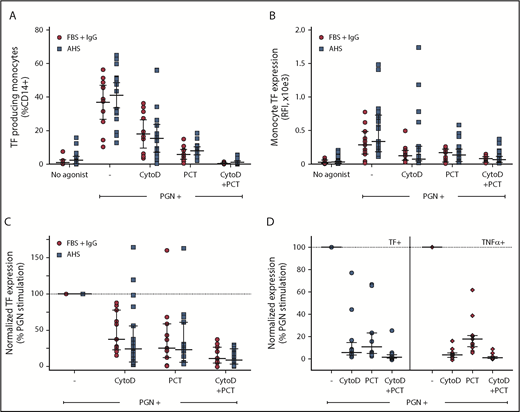
PGN-induced monocyte TF expression is mediated by cell surface and internalization-dependent signaling. (A) The frequency of PGN-induced procoagulant monocytes is reduced by the phagocytic inhibitor cytoD and/or Syk inhibition proximally downstream of FcγR signaling with piceatannol (PCT). (B) TF fluorescence intensity in PGN-stimulated monocytes is sensitive to cytoD and/or PCT. (C) Pairwise normalization of monocyte TF expression after PGN stimulation in the presence of cytoD and/or PCT. (A-C) Similar results were obtained in the presence of autologous donor serum (AHS; blue squares) or fetal bovine serum supplemented with human IgG (FBS + IgG; red circles). (D) Comparison of relative sensitivity to cytoD and/or PCT of PGN-induced paired procoagulant (TF+) and proinflammatory (TNF-α+) monocyte responses. Individual data represent inducible marker expression, either TF or TNF-α, in the presence of inhibitors after normalization to PGN-induced expression in the absence of pharmacologic inhibitors. Horizontal bars depict median and IQR in all panels. Graphic representation of comparisons between groups has been omitted for clarity and is summarized in text.
PGN-induced monocyte TF expression is mediated by cell surface and internalization-dependent signaling. (A) The frequency of PGN-induced procoagulant monocytes is reduced by the phagocytic inhibitor cytoD and/or Syk inhibition proximally downstream of FcγR signaling with piceatannol (PCT). (B) TF fluorescence intensity in PGN-stimulated monocytes is sensitive to cytoD and/or PCT. (C) Pairwise normalization of monocyte TF expression after PGN stimulation in the presence of cytoD and/or PCT. (A-C) Similar results were obtained in the presence of autologous donor serum (AHS; blue squares) or fetal bovine serum supplemented with human IgG (FBS + IgG; red circles). (D) Comparison of relative sensitivity to cytoD and/or PCT of PGN-induced paired procoagulant (TF+) and proinflammatory (TNF-α+) monocyte responses. Individual data represent inducible marker expression, either TF or TNF-α, in the presence of inhibitors after normalization to PGN-induced expression in the absence of pharmacologic inhibitors. Horizontal bars depict median and IQR in all panels. Graphic representation of comparisons between groups has been omitted for clarity and is summarized in text.
Distribution of monocyte immune responses to PGN
Monocyte immune responses to PGN include both proinflammatory and procoagulant outcomes. We investigated the coexpression of TF and TNF-α after PAMP stimulation. As shown in Figure 4, the immune outcomes in PGN-activated monocytes split evenly between single procoagulant (22.5% TF+TNF-α–) and double procoagulant/proinflammatory (22.2% TF+TNF-α+) monocytes, whereas single proinflammatory cells were half represented (13.4% TF–TNF-α+). In contrast, TLR signaling initiated by either LPS or Pam3CSK4 preferentially induced single procoagulant responsive monocytes (44.3% and 36.1% TF+TNF-α– responsive monocytes, respectively) and fewer double procoagulant/proinflammatory monocytes (17.3% and 6.5% TF+TNF-α+). Heat-killed B anthracis was the most potent stimulus and induced a higher representation of double procoagulant/proinflammatory monocytes (47.7% TF+TNFα+) than single procoagulant (18.4% TF+TNF-α–) or single proinflammatory (17.1% TF–TNF-α+) monocytes. Overall, our data indicate that monocyte procoagulant and proinflammatory responses to PGN use the same signaling avenues and produce overlapping subpopulations. Furthermore, PGN activation of monocytes models the outcomes of gram-positive stimulation better than TLR stimulation.
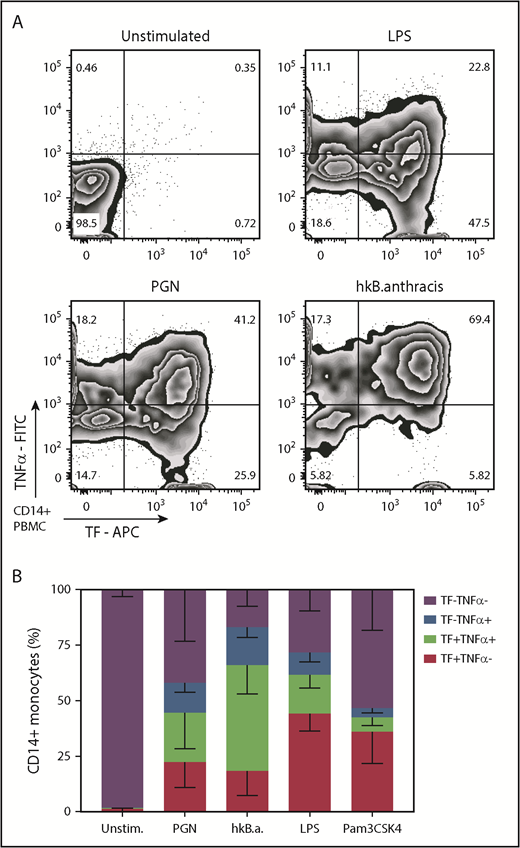
PAMPs induce overlapping procoagulant and proinflammatory responses in human monocytes. PBMCs were stimulated for 6 hours with PGN (10 µg/mL), heat-killed (hk) B anthracis (107 CFU/mL), LPS (1 µg/mL), or Pam3CSK4 (1 µg/mL) in the presence of BFA before intracellular quantification of TF and TNF-α by flow cytometry. (A) Representative distribution of procoagulant (TF+) and proinflammatory (TNF-α+) responses in PAMP-stimulated monocytes. (B) Average distribution of procoagulant and proinflammatory responses in PAMP-stimulated monocytes in the cohort studied (n = 11). Column segments represent mean and standard deviation for each population depicted in the legend. FITC, fluorescein isothiocyanate.
PAMPs induce overlapping procoagulant and proinflammatory responses in human monocytes. PBMCs were stimulated for 6 hours with PGN (10 µg/mL), heat-killed (hk) B anthracis (107 CFU/mL), LPS (1 µg/mL), or Pam3CSK4 (1 µg/mL) in the presence of BFA before intracellular quantification of TF and TNF-α by flow cytometry. (A) Representative distribution of procoagulant (TF+) and proinflammatory (TNF-α+) responses in PAMP-stimulated monocytes. (B) Average distribution of procoagulant and proinflammatory responses in PAMP-stimulated monocytes in the cohort studied (n = 11). Column segments represent mean and standard deviation for each population depicted in the legend. FITC, fluorescein isothiocyanate.
Procoagulant responses to PGN are sensitive to brefeldin A
To assess proinflammatory outcomes of PGN stimulation, we used brefeldin A (BFA; 3 µg/mL) to block cytokine secretion (Figure 5). Despite being a transmembrane protein, TF can be released from the monocyte surface by vesiculation and formation of TF-laden microparticles.47 BFA inhibition of the secretory pathway was expected to trap TF intracellularly and reduce microparticle loss of TF from the monocyte surface. As hypothesized, BFA treatment prevented cell surface exposure with a concomitant increase in the intracellular TF pool after LPS stimulation. BFA, however, did not block TF release from LPS-activated cells (supplemental Figure 2), suggesting expulsion of TF-laden exosomes independent of cell surface TF trafficking. Overall, BFA increased TF antigen expression in almost all (24 of 26) LPS-stimulated PBMCs and similarly enhanced the release of TF from activated cells. In contrast, BFA reduced TF expression in 69% (18 of 26) of the paired PGN-stimulated PBMCs (P < .0001, compared with LPS-stimulated PBMCs; RM 1-way ANOVA). TF antigen levels were ∼15% lower in PGN-stimulated PBMCs in the presence vs the absence of BFA (P = .0145, ratio-paired Student t test). Similarly, BFA treatment reduced the release of TF from PGN-stimulated monocytes (P = .0059, ratio-paired Student t test). The overall decrease in both the intracellular TF pool and the release of TF from PGN-activated monocytes suggests that procoagulant monocyte responses to PGN might be amplified by soluble mediators secreted by PGN-stimulated PBMCs and blocked by BFA.
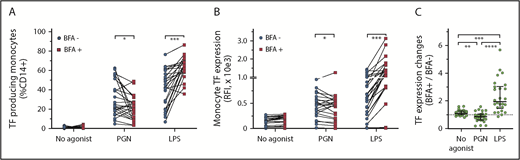
Differential effect of BFA on TF expression in PAMP-stimulated monocytes. (A) Pairwise representation of BFA treatment on procoagulant responses after PGN (10 µg/mL) or LPS (1 µg/mL) stimulation. BFA increases the fraction of procoagulant monocytes (TF+CD14+) after LPS stimulation but decreases procoagulant monocytes after PGN stimulation. (B) Pairwise representation of the differential effect of BFA on monocyte TF fluorescence intensity after PGN or LPS stimulation. (C) Pairwise comparison of normalized changes in PAMP-induced monocyte TF intensity (TF geometric mean BFA+/TF geometric mean BFA–) in the studied cohort (n = 26). Differences between paired groups were analyzed by using ratio-paired Student t tests (panels A and B) or RM 1-way ANOVA (panel C).
Differential effect of BFA on TF expression in PAMP-stimulated monocytes. (A) Pairwise representation of BFA treatment on procoagulant responses after PGN (10 µg/mL) or LPS (1 µg/mL) stimulation. BFA increases the fraction of procoagulant monocytes (TF+CD14+) after LPS stimulation but decreases procoagulant monocytes after PGN stimulation. (B) Pairwise representation of the differential effect of BFA on monocyte TF fluorescence intensity after PGN or LPS stimulation. (C) Pairwise comparison of normalized changes in PAMP-induced monocyte TF intensity (TF geometric mean BFA+/TF geometric mean BFA–) in the studied cohort (n = 26). Differences between paired groups were analyzed by using ratio-paired Student t tests (panels A and B) or RM 1-way ANOVA (panel C).
Procoagulant responses to PGN are amplified by proinflammatory cytokines
To investigate the paracrine amplification of procoagulant signals after PGN stimulation, we performed supernatant transfer studies. Donor PBMCs (Figure 6A) were stimulated with PGN, and soluble mediators released in the supernatants (conditioned media) were transferred to recipient, PGN-naive PBMCs. PGN induced procoagulant responses in donor PBMCs in a time-dependent manner and similarly promoted the release of soluble mediators that transferred the procoagulant potential to naive cells. These mediators require viable donor cells, as paraformaldehyde fixation of donor PBMCs before the PGN treatment abolished the procoagulant signal transfer. Interestingly, we observed segregation of donor responses after short-term PGN stimulation (2 and 4 hours). Two-thirds of the study group (7 of 11 donors, fast responders) secreted procoagulant mediators within the first 2 hours of PGN stimulation, whereas the remainder (4 of 11 donors, slow responders) required the full 6-hour time-course for release. One donor contained procoagulant mediators in their serum, which stimulated recipient-naive PBMCs; PGN stimulation of this donor further increased the transferred procoagulant signals.

PGN-induced procoagulant responses are amplified by cytokine signaling. (A) Procoagulant signals from PGN-stimulated donor PBMCs (blue circles) can be transferred to PGN-naive recipient PBMCs (red squares) by soluble mediators through paracrine signaling. Individual (symbols) and median responses (columns) are illustrated, depicting frequency of procoagulant monocytes (TF+CD14+; left panel) or monocyte TF fluorescence intensity (right panel). (B) Procoagulant responses induced by PGN (10 µg/mL) are sensitive to neutralization of paracrine and autocrine cytokine signaling. Pairwise representation of cytokine neutralization effect on the frequency of procoagulant monocytes induced by PGN (left panel) and normalized changes in PGN-induced monocyte TF antigen expression (right panel) in the presence of neutralizing antibodies are shown.
PGN-induced procoagulant responses are amplified by cytokine signaling. (A) Procoagulant signals from PGN-stimulated donor PBMCs (blue circles) can be transferred to PGN-naive recipient PBMCs (red squares) by soluble mediators through paracrine signaling. Individual (symbols) and median responses (columns) are illustrated, depicting frequency of procoagulant monocytes (TF+CD14+; left panel) or monocyte TF fluorescence intensity (right panel). (B) Procoagulant responses induced by PGN (10 µg/mL) are sensitive to neutralization of paracrine and autocrine cytokine signaling. Pairwise representation of cytokine neutralization effect on the frequency of procoagulant monocytes induced by PGN (left panel) and normalized changes in PGN-induced monocyte TF antigen expression (right panel) in the presence of neutralizing antibodies are shown.
The most likely procoagulant mediators are proinflammatory cytokines such as TNF-α, IL-1β, and IL-6, important amplifiers of clotting dysregulation in sepsis models, including anthrax. Neutralizing antibodies to either TNF-α or IL-1β reduced both the frequency of TF+CD14+ monocytes as well as overall monocyte TF antigen expression in 70% to 85% of the studied donors (P = .0398 and P = .0094, respectively; ratio paired Student t test compared with PGN-stimulated PBMCs) (Figure 6B). In contrast, IL-6 blockade showed reduction of procoagulant responses in a small fraction of donors and as such was not statistically significant. The most effective reduction of procoagulant responses in our study cohort was obtained by combined neutralization of TNF-α and IL-1β (median [IQR] reduction of 47% [42%-72%]; P < .0001, ratio paired Student t test vs PGN-stimulated PBMCs). Cytokine neutralization did not reduce procoagulant responses to LPS (supplemental Figure 3). Overall, our data indicate that primary procoagulant responses to PGN are amplified by a positive feedback mediated by TNF-α and IL-1β proinflammatory cytokines.
Discussion
We recently described the procoagulant potential of PGN in a late-stage anthrax sepsis model in nonhuman primates.30 Both coagulation pathways were initiated, and we observed TF-expressing monocytes in circulation. In the current study, we investigated the mechanism by which PGN induces monocyte TF expression using a large cohort of healthy volunteers. Our data show that: (1) PGN is a potent inducer of monocyte TF similar to LPS, the main procoagulant PAMP in gram-negative sepsis; (2) in vitro, monocyte responses to PGN model gram-positive outcomes better than TLR agonists; (3) monocyte procoagulant and proinflammatory responses to PGN use the same signaling avenues, either internalization-dependent and/or surface FcγR signaling; and (4) primary procoagulant responses to PGN signaling are reinforced by proinflammatory feedback through TNF-α and IL-1β.
TF, the receptor and allosteric cofactor of plasma protease FVII, is the main initiator of blood coagulation during both hemostatic and thrombotic events.3 Similar to previous studies,5 our study cohort shows that basal TF expression in circulating human monocytes is uncommon, and when it happens, it is transient and likely due to unscreened physiologic challenges. These low levels of basal TF antigen were mainly detected after cellular permeabilization, thus intracellularly and spatially segregated from contact with circulating factors.
Intravascular TF can be induced under pathologic conditions (eg, bacterial or viral infections, systemic inflammatory conditions in autoimmune disease, cancer) and promotes thrombotic and proinflammatory manifestations.48 LPS is the main procoagulant PAMP in gram-negative infections and can induce intravascular TF expression8,15 directly through TLR4 signaling49 or secondary to the inflammatory response.50,51 Despite a similar incidence in DIC, the primary procoagulant PAMP in gram-positive infections has been elusive and assumed to be TLR2-driven.52 In experimental anthrax, however, severe coagulopathies are induced by challenges with cell wall PGN30,31 with no TLR agonist ability.37 Similarly, PGN induced monocyte TF expression in all healthy volunteers tested in the current study. Due to its abundant representation in the cell wall of gram-positive pathogens, PGN was hypothesized to be the main procoagulant PAMP in gram-positive sepsis. Despite engaging distinct signaling networks, PGN and LPS procoagulant responses were fairly similar in the study cohort, although we observed individuals who preferentially reacted to one PAMP or the other. Furthermore, the amplitude of procoagulant responses to both LPS and PGN suggests engagement of the classical CD14+CD16– monocytes, which were recently shown to be the primary procoagulant monocytes during viral infection.53
Monocyte procoagulant and proinflammatory responses to PGN overlapped in all donors tested and were better at resembling the responses to B anthracis bacterium than TLR signaling. In contrast to PGN, which equally promoted double procoagulant/proinflammatory (TF+TNF+) and single procoagulant (TF+TNF–) monocytes, TLR signaling preferentially induced single procoagulant (TF+TNF–) monocytes under the study conditions. This difference may be due to different immune functions within the monocyte subsets as described elsewhere.54 Mukherjee et al proposed that classical CD14+CD16– monocytes have an augmented immune-processing role due to enhanced expression of antigen-presenting molecules, whereas the intermediary (CD14+CD16+) and nonclassical (CD14dimCD16+) monocytes have an augmented inflammatory role due to enhanced expression of TLRs. As such, LPS stimulation will promote procoagulant responses in classical monocytes, as described recently,53 leading to single procoagulant monocytes (TF+TNF–) while preferentially inducing proinflammatory responses (TF–TNF+) in nonclassical monocytes. Lower TLR expression in classical monocytes will lead to slower proinflammatory signaling and subsequent conversion of single procoagulant into double procoagulant/proinflammatory classical monocytes (TF+TNF+). In contrast, immune recognition of PGN is expected to preferentially occur in classical monocytes due to their augmented antigen-processing role. As such, both proinflammatory and procoagulant outcomes are initiated in the same monocyte subset, leading to similar representation of outcomes.
Immune responses to cell wall PGN require recognition by immune receptors,42 internalization and lysosomal processing of PGN,34,35 cytosolic release of monomeric PGN subunits, and subsequent activation of NOD intracellular sensors38,39 and/or inflammasome.40 Immunoglobulin FcγRs are critical for efficient processing of PGN opsonized by IgG or serum amyloid P,36,42 and complement opsonization of PGN has also been observed41,55 and may contribute to PGN phagocytosis. In addition, surface-exposed CD14 binds PGN and contributes to PGN-induced monocyte responses.56,57 Although CD14 coreceptor function during LPS-TLR4 activation has been detailed,58,59 it is unknown to which receptor CD14 shuttles PGN (although we suspect to aid recognition by low-affinity IgG receptors). Due to PGN repetitive structure and size, IgG opsonization of PGN is expected to cluster FcγRs, thus supporting FcγR-signaling platforms at the cell surface, which critically requires Syk tyrosine kinase function44 for both surface signaling and phagocytosis. PGN internalization may occur through phagocytosis or micropinocytosis, and both pathways require reorganization of actin cytoskeleton. At high concentrations, we observed PGN internalization in the absence of serum opsonization, indicative of macropinocytosis, and monocyte PGN responses were sensitive to amiloride, the most common pharmacologic inhibitor of macropinocytosis.60 Nevertheless, amiloride side effects, such as decreased phosphatidylinositol 3-kinase signaling61 and abnormal cellular distribution and function of lysosomes,62 could potentially affect surface FcγR signaling as well as internalization-dependent PGN signaling.63 We used cytoD, an inhibitor of actin polymerization,64 in the current study to block PGN internalization by both receptor-mediated phagocytosis and macropinocytosis. After cytoD blockade, residual PGN procoagulant responses were sensitive to piceatannol, a Syk kinase inhibitor that blocks FcγR signaling.65,66 Thus, we show for the first time that engagement of surface FcγR immune receptors leads to monocyte TF expression. Interestingly, the procoagulant responses to PGN were less sensitive to cytoD inhibition than the paired proinflammatory responses assessed in parallel. This finding could indicate that surface FcγR engagement preferentially promotes TF expression, whereas internalization-dependent signaling promotes both proinflammatory and procoagulant responses. The procoagulant and proinflammatory outcomes to PGN make use of the same signaling avenues; to date, we are unable to identify pharmacologic inhibitors that selectively affect only one or the other. This scenario suggests an intimate juxtaposition of procoagulant and proinflammatory responses in PGN-stimulated monocytes but raises the intriguing question as to why some monocytes exhibit single responses while others show a double procoagulant/proinflammatory phenotype. These populations may reflect variable signaling kinetics in individual cells, or there may be intrinsic switches that facilitate modulation of the biological outcomes. Further detailed molecular analysis of these signaling pathways will help elucidate these differences.
Activated monocytes release cell membrane TF through vesiculation,47 and TF-positive monocyte-derived microparticles promote thrombosis in multiple pathologies.67 Activation of inflammasome, which also detects acetylated PGN sugars,40 stimulates TF microparticle release from macrophages.68 BFA inhibition of the conventional secretory pathway blocks TF exposure at the cell surface and promotes intracellular TF accumulation in LPS-stimulated monocytes. Unexpectedly, BFA potentiated the release of TF from LPS-stimulated cells independent of plasma membrane exposure of TF. In contrast to LPS, BFA decreased both intracellular accumulation and TF release from PGN-activated monocytes. BFA-mediated decrease of TF+CD14+ fractions suggested a paracrine amplification of PGN procoagulant responses by soluble mediators, which we unequivocally proved by supernatant transfer studies. In fact, segregation of responses after short-term PGN stimulation (2 hours) showed that fast responders (7 of 11 [64% of donors]) released procoagulant mediators early, even before significant translation of TF by these PBMCs. These early secreted mediators could potentiate monocyte TF expression through an autocrine mechanism as indicated by cytokine neutralization assays and decreased antigen levels in BFA-treated cells.
Proinflammatory cytokines, mainly TNF-α, IL-1β, and IL-6, enhance the coagulopathy associated with endotoxemia and sepsis.69 Procoagulant cytokines are elevated in both gram-negative and gram-positive human sepsis patients,70 as well as in experimental sepsis models, including nonhuman primates challenged with B anthracis27 and anthrax PGN.30 Within PBMCs, monocytes drive the proinflammatory responses to PGN,34 and, in this study, we report a partial overlap of TF+ and TNF-α+ monocytes. It is thus likely that proinflammatory cytokines secreted by PGN-stimulated monocytes potentiate procoagulant responses by an autocrine mechanism. Using neutralizing antibodies against procoagulant cytokines (eg, TNF-α, IL-1β) or cytokine receptors (eg, IL-6R), we found that PGN procoagulant responses are potentiated by TNF-α and IL-1β feedback signaling and less so by IL-6R. Both IL-1β and TNF-α blockade, by themselves but more so in combination, significantly reduced the frequency of procoagulant monocytes and TF antigen levels, indicating both an autocrine and paracrine amplification of the procoagulant responses. Neutralizing the amplification of TF expression by dual inhibition of TNF-α and IL-1β could prove beneficial in reducing the coagulopathy in gram-positive sepsis without affecting the primary immune response to PGN and possibly the invading pathogen.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors are grateful to all volunteers who self-enrolled in this study. They thank Florea Lupu, Oklahoma Medical Research Foundation, for his critical review of the manuscript.
This work was supported by grants from the National Institutes of Health, National Institute of Allergy and Infectious Diseases (U19AI062629 and 1R21 AI113020) (K.M.C.)
Authorship
Contribution: N.I.P. performed the experiments and analyzed the data; A.G., T.B., and K.L. purified and characterized the PGN; T.B. and K.L. collected blood from donors, managed the donor database, and provided de-identified samples for experiments; N.I.P. and K.M.C. designed the study and wrote the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for A.G. is Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC.
Correspondence: Narcis Ioan Popescu, Oklahoma Medical Research Foundation, 825 NE 13th St, Oklahoma City, OK 73104; e-mail: narcis-popescu@omrf.org.

