

Key Points
CB-VSTs can be feasibly generated and are safe.
CB-VSTs persist long term after infusion in CBT recipients.

Abstract
Adoptive transfer of virus-specific T cells (VSTs) has been shown to be safe and effective in stem cell transplant recipients. However, the lack of virus-experienced T cells in donor cord blood (CB) has prevented the development of ex vivo expanded donor-derived VSTs for recipients of this stem cell source. Here we evaluated the feasibility and safety of ex vivo expansion of CB T cells from the 20% fraction of the CB unit in pediatric patients receiving a single CB transplant (CBT). In 2 clinical trials conducted at 2 separate sites, we manufactured CB-derived multivirus-specific T cells (CB-VSTs) targeting Epstein-Barr virus (EBV), adenovirus, and cytomegalovirus (CMV) for 18 (86%) of 21 patients demonstrating feasibility. Manufacturing for 2 CB-VSTs failed to meet lot release because of insufficient cell recovery, and there was 1 sterility breach during separation of the frozen 20% fraction. Delayed engraftment was not observed in patients who received the remaining 80% fraction for the primary CBT. There was no grade 3 to 4 acute graft-versus-host disease (GVHD) associated with the infusion of CB-VSTs. None of the 7 patients who received CB-VSTs as prophylaxis developed end-organ disease from CMV, EBV, or adenovirus. In 7 patients receiving CB-VSTs for viral reactivation or infection, only 1 patient developed end-organ viral disease, which was in an immune privileged site (CMV retinitis) and occurred after steroid therapy for GVHD. Finally, we demonstrated the long-term persistence of adoptively transferred CB-VSTs using T-cell receptor-Vβ clonotype tracking, suggesting that CB-VSTs are a feasible addition to antiviral pharmacotherapy.
Introduction
Over the last 25 years, adoptive immunotherapy using virus-specific T cells (VSTs) has emerged as a safe and effective alternative to antiviral pharmacotherapy.1-5 Although traditional antiviral therapies for patients after bone marrow or cord blood transplant (CBT) can be costly, ineffective, and often associated with myelosuppression and renal failure, antiviral T-cell infusions (currently in pivotal phase 3 trials) are safe, effective, and have long-term persistence in vivo.6-11 One limitation of ex vivo generation of antiviral T cells is the need for the donor to have had prior exposure to the virus, which renders the approach unsuitable for CBT when the graft T cells are virus naïve.12 Third-party, off-the-shelf allogeneic T cells are effective for patients who cannot tolerate conventional antiviral pharmacotherapy,13,14 but studies have shown that persistence of these cells is sustained for only approximately 12 weeks.15 Hence, third-party VSTs may not provide the required long-term protection after CBT when full immune reconstitution can be delayed for as long as 2 years. It would therefore be advantageous to develop a donor-derived CB-VST product to infuse after CBT to rapidly restore long-term immune reconstitution in high-risk patients.16-20
CB contains predominantly naïve T cells,21 which makes the ex vivo generation of VSTs difficult and thus delays translation to the bedside. Sun et al22 and Park et al23 reported the generation of Epstein-Barr virus (EBV)– and cytomegalovirus (CMV)-specific T cells from CB but not in sufficient doses for clinical application. In 2009, we published the first Good Manufacturing Practice–applicable approach to manufacturing CMV-, EBV-, and adenovirus-specific T cells from CB using 20% of a fractionated CB unit.12,24 However, because low CD34+ cell counts in CB units contribute to delayed engraftment when compared with bone marrow or peripheral blood stem cell transplants,25 it was not known whether infusing only 80% of the CBT might cause delayed engraftment, which makes this strategy unfeasible. Moreover, the safety and efficacy of adoptively transferred naïve-derived T cells have been largely studied in murine models.26,27 Thus, in contrast to the well-established safety and efficacy of memory-derived VSTs from peripheral blood, clinical trials using human naïve–derived T cells are sparse, and little is known about the long-term safety and efficacy of CB-derived VSTs (CB-VSTs).
Previously, we showed that by using professional antigen-presenting cells (APCs) and cytokines to mimic in vivo priming conditions of naïve T cells, we could expand CMV-, EBV-, and adenovirus-specific T cells from CB and CMV-naïve adult donors.28 Interestingly, these cells recognized atypical epitopes of the CMV immunogenic antigen pp65,12 and it was unknown whether T cells derived from the naïve population that recognized atypical epitopes would be effective. On the basis of our preclinical data, we hypothesized that (1) the 20% fraction derived from the whole CB unit transplanted to the patient could be used to expand VSTs, (2) the use of this fraction from adequately sized CB units would not delay engraftment, and (3) despite their atypical epitope recognition, VSTs from CB could effectively treat or prevent viral infections after CBT.
To test these hypotheses, we established a clinical trial entitled “Adoptive Transfer of Cord Blood T Cells to Prevent and Treat CMV and Adenovirus Infections after Transplantation” (ACT-CAT). Four years later, ACT-CAT2 was initiated with modified manufacturing to eliminate gene-modified APCs by using overlapping peptide libraries targeting 2 immunodominant antigens from each of the 3 targeted viruses (CMV, EBV, and adenovirus). We showed that adoptive transfer of CB-derived T cells to 14 patients after CBT was safe and effective in preventing viral disease in the highest-risk patients and that the infused cells could persist long term as evaluated by using T-cell receptor-Vβ (TCR-Vβ) sequencing.
Methods
Patient eligibility
Patients with either malignant or nonmalignant disease undergoing 4/6-6/6 HLA matched unrelated CBT and at high risk for or with CMV, EBV, or adenovirus reactivation or disease were eligible for enrollment in ACT-CAT (NCT00880789) and ACT-CAT2 (NCT01923766) at the participating institutions. Risk for CMV, EBV, or adenovirus reactivation was determined by the treating physician after considering history of viral exposure or seropositivity, use of serotherapy in the conditioning regimen, and history of immunodeficiency. All the transplant patients were recipients of a single CB graft. All patients had CB units that were formatted into 2 fractions with the transplant component (80% fraction) containing a minimum of 2.5 × 107 total nucleated cells per patient weight in kilograms (TNCs/kg). The remaining 20% fraction was used for VST generation. Eligibility for VST infusion required an absolute neutrophil count >0.5 × 109/L with predominant donor cell chimerism, no grade 2 to 4 acute graft-versus-host disease (GVHD), and receipt of ≤0.5 mg/kg per day prednisone equivalent systemic steroid therapy at the time of enrollment. Patients with primary disease relapse were not eligible.
Generation of CB-VST cell cultures
CB-VSTs were generated from licensed or unlicensed frozen CB units under an investigational new drug application (IND) from National Marrow Donor Program CB banks. For patients enrolled on the ACT-CAT clinical trial at Baylor College of Medicine or ACT-CAT2 at Children’s National Medical Center, the 80% fraction of the clinical CB unit was infused in the patient as specified by the transplantation protocol. The 20% fraction of the unit was used to manufacture CB-VSTs according to the INDs with the US Food and Drug Administration and as detailed below and in Figure 1. The INDs held at Baylor College of Medicine and Children’s National Medical Center were reviewed by the Food and Drug Administration and approved by the institutional review boards of each respective institution. Participants or guardians gave informed consent upon enrollment.
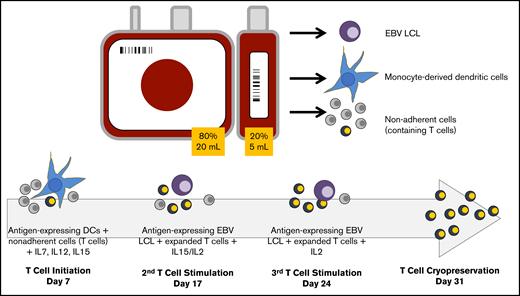
Schematic of CB-VST manufacture in ACT-CAT and ACTCAT2 clinical trials. Eighty percent of the CB unit was used as the CB transplant graft, and 20% was used to expand the VST. From the 20% fraction, 5 to 10 million peripheral blood mononuclear cells (PBMCs) were used for EBV lymphoblastoid cell line (LCL) generation; the remaining cells were used to generate dendritic cells (DCs) or used as the source of T cells. DCs were matured and transduced with the Ad5f35-pp65 vector (ACT-CAT) on day 5 and/or cocultured with overlapping viral peptides (PepMix) (ACT-CAT2), then cocultured with the nonadherent fraction (containing T cells) on day 7 in the presence of IL-7, IL-12, and IL-15. At the second and third stimulation, EBV-transformed B cells were transduced with the same Ad5f35-pp65 vector 2 days before the T-cell stimulation or pulsed with PepMix the day of stimulation, which was performed in the presence of IL-15 (second stimulation) and IL-2 (third stimulation).
Schematic of CB-VST manufacture in ACT-CAT and ACTCAT2 clinical trials. Eighty percent of the CB unit was used as the CB transplant graft, and 20% was used to expand the VST. From the 20% fraction, 5 to 10 million peripheral blood mononuclear cells (PBMCs) were used for EBV lymphoblastoid cell line (LCL) generation; the remaining cells were used to generate dendritic cells (DCs) or used as the source of T cells. DCs were matured and transduced with the Ad5f35-pp65 vector (ACT-CAT) on day 5 and/or cocultured with overlapping viral peptides (PepMix) (ACT-CAT2), then cocultured with the nonadherent fraction (containing T cells) on day 7 in the presence of IL-7, IL-12, and IL-15. At the second and third stimulation, EBV-transformed B cells were transduced with the same Ad5f35-pp65 vector 2 days before the T-cell stimulation or pulsed with PepMix the day of stimulation, which was performed in the presence of IL-15 (second stimulation) and IL-2 (third stimulation).
Generation of dendritic cells from CB mononuclear cells
To generate the source of APCs for the first T-cell stimulation, CB was thawed at 37°C, centrifuged at 450g for 10 minutes, and resuspended in RPMI 1640. CB mononuclear cells (CBMCs) were then isolated by Ficoll (Lymphoprep; Nycomed, Oslo, Norway) gradient separation. CBMCs were washed twice, resuspended in CellGenix media (CellGenix USA, Antioch, IL), and plated at approximately 1 × 107 cells per mL in 2 mL of dendritic cell (DC) medium (CellGenix media plus 2 mM L-glutamine) (GlutaMAX; Invitrogen, Carlsbad, CA) in a 6-well plate (Costar, Corning, NY) for 1 to 2 hours at 37°C in a humidified CO2 incubator. Nonadherent cells were removed by rinsing with 1× phosphate-buffered saline (Sigma, St. Louis, MO) and cryopreserved for later use. Cells adherent after 1 to 2 hours were cultured in DC media with 800 U/mL granulocyte-macrophage colony-stimulating factor (Sargramostim Leukine; Immunex, Seattle, WA) and 500 U/mL interleukin-4 (IL-4) (R&D Systems, Minneapolis, MN) for 5 days. On day 5, the cells were matured as described below.
Maturation of DCs and transduction with Ad5f35-pp65 vector (ACT-CAT)
CB-derived DCs were transduced with a chimeric adenoviral vector expressing the immunodominant CMVpp65 antigen (Ad5f35-pp65)29 at a multiplicity of infection of 1000 viral particles on day 512 and matured using a cytokine cocktail consisting of 800 U/mL granulocyte-macrophage colony-stimulating factor, 1000 U/mL IL-4, 10 ng/mL IL-1β, 10 ng/mL tumor necrosis factor-α, 100 ng/mL IL-6 (R&D Systems), and 1 μg/mL prostaglandin E1 (Sigma) (cytokine cocktail) for 1 to 2 days. On days 6 to 7, the DCs were harvested and used to stimulate T cells present in the cryopreserved nonadherent fraction.
Maturation and peptide pulsing of DCs (ACT-CAT2)
CB DCs were generated as described above, but instead of adenoviral vector transduction, they were pulsed with viral PepMix libraries containing overlapping 15mer peptides (JPT, Berlin, Germany) encompassing viral antigens IE-1 and pp65 (CMV), LMP2 and EBNA1 (EBV), and hexon and penton (adenovirus).30 At the time of maturation, the same maturation cocktail was used as above but with the addition of 30 ng/mL lipopolysaccharide. On days 6 to 7, the mature DCs were harvested, centrifuged at 400g for 5 minutes, pulsed with 100 ng peptide master mix per 10 million DCs for 30 to 60 minutes, and then plated in T-cell medium containing human serum in a 24-well plate at approximately 1 to 2 × 105 cells per well.
Generation and adenoviral transduction of EBV-transformed B-cell lines from CBMCs
Because CB-VSTs are generated from a naïve T-cell donor source, multiple stimulations are needed to ensure sufficient cell yield and specificity.12,28 CB-derived DCs were required for T-cell priming (first stimulation), and lymphoblastoid cell lines (LCLs) were used to expand the CB-derived VSTs (second and third stimulations). To generate LCLs, 5 × 106 CBMCs were infected at the time of DC plating with supernatants from an EBV-producing B95-8 working cell bank.3 Once the LCLs were established, sufficient LCLs were either transduced with the Ad5f35-pp65 vector (ACT-CAT)12 or pulsed with overlapping peptide libraries (ACT-CAT2) as previously described.31
T-cell initiation and stimulations
At the time of initiation, the nonadherent cells that were collected during the generation of DCs were thawed and washed, and 2 million nonadherent cells were added to each well of a 24-well plate for a T-cell:DC ratio of approximately 1:10 to 1:20. Then, 10 ng/mL IL-7, 10 ng/mL IL-12, and 5 ng/mL IL-15 were added to the T-cell media–containing human serum. After 9 to 12 days, the T cells were again cryopreserved until the EBV LCLs were sufficiently expanded. T cells were then added to either 24-well plates at a T-cell:LCL ratio of 4:1 or to Grex10 devices at 5:1 in the presence of 5 ng/mL IL-15. This was again repeated for the third stimulation, with IL-2 being substituted for IL-15. After the second stimulation, cells were fed every 3 to 4 days with 50 to 100 U/mL IL-2.
Product release
All T cells were released under a certificate of analysis after appropriate testing was completed. Release testing was as follows: <2% CD3–/CD83+; <2% CD19; >70% viable by Trypan blue; negative for Mycoplasma; <5.0 EU/mL endotoxin; sterile for anaerobic/aerobic bacteria at 4 days or 21 days, sterile for fungus at 21 or 28 days; and <10% killing of patient or related-donor phytohemagglutinin blasts in a chromium-release cytotoxicity assay.
Enzyme-linked immunospot assay
Enzyme-linked immunospot (ELISPOT) analysis was used to test the frequency of CB-VSTs secreting interferon-γ (IFN-γ) when stimulated with PepMixes for CMV, EBV, and adenovirus as previously described.12 Spot-forming cells (SFCs) were enumerated by Zellnet Consulting (New York, NY) and compared with input cell numbers to obtain the frequency of virus-reactive T cells.
Immunophenotyping
CB-VSTs were stained and analyzed with monoclonal antibodies to CD3, CD4, CD8, CD56/16, CD19, CD83, CD45RA, and CD62L (Becton Dickinson, Franklin Lakes, NJ and Miltenyi Biotec).
TCR sequencing
Frozen peripheral blood samples were obtained from patients enrolled on ACT-CAT or ACT-CAT2. In some cases, cells were sorted using magnetic-activated cell sorting selection based on expression of CD45RA. TCR-Vβ sequencing was performed using the ImmunoSEQ platform from genomic DNA with primers specific for 45 expressed Vβ regions and 13 Jβ regions (Adaptive Biotechnologies, Seattle, WA). In most cases, bulk cytotoxic T lymphocyte products or patient follow-up peripheral blood mononuclear cell (PBMC) samples were analyzed by Survey level (VST) or deep T-cell receptor (PBMC) sequencing. Bioinformatic analysis was performed on the sequencing data using the ImmunoSEQ platform.32
Clinical trial design and statistical analysis
The 2 phase 1 dose-escalation trials were designed to evaluate the safety of CB-VSTs with dose escalation guided by the modified continual reassessment method to determine the maximum tolerated dose of VSTs.33 Dose-limiting toxicity (DLT) was defined as the development of National Cancer Institute grade 3 to 4 nonhematologic toxicity that was attributed to the VSTs within 30 days or GVHD within 45 days of infusion. Maximum tolerated dose was defined as the dose at which the probability of DLT is at most 21%. For the phase 1 trial, data analyses were mainly descriptive. Comparisons between groups were performed in GraphPad PRISM 8.01 using a 2-tailed Student t test assuming equal variance. P < .05 was considered statistically significant.
Results
Patient and transplant characteristics
Patient demographics and transplant characteristics of the 14 patients (9 on ACT-CAT and 5 on ACT-CAT2) who received a CB-VST infusion are shown in Table 1. The median age was 23 months (range, 12 months to 8 years), and an equal number of patients had an underlying malignant or nonmalignant disorder as the indication for CBT. No patients on ACT-CAT received alemtuzumab or antithymocyte globulin before transplantation, whereas 3 of 5 patients in ACT-CAT2 did. Most patients (n = 13) received cyclosporine and mycophenolate mofetil as part of their GVHD prophylaxis, and 11 patients received steroids after transplantation but before CB-VST infusion as GVHD prophylaxis or treatment of GVHD or engraftment syndrome after the CBT. Four patients had a 6/6 HLA match (antigen matched for HLA-A and B, allele matched for HLA-DRB1), 9 had a 5/6 match, and 1 had a 4/6 match. All patients underwent transplantation using a CB unit from a single unrelated donor.
Demographics of patients receiving CB-derived VST infusions
| Patient ID | Diagnosis | Age, y | Sex | Weight, kg | Conditioning regimen | HLA match (of 6) | Immunosuppression before VST | Recipient CMV serostatus | TNCs from UCB, × 107 | TNCs/kg of UCB fraction used for primary transplant |
|---|---|---|---|---|---|---|---|---|---|---|
| ACT-CAT | ||||||||||
| P2891 | Fanconi anemia | 5 | M | 13.0 | Flu/Cy/TBI | 5 | CSA,MMF, steroids | Positive | 161 | 9.9 |
| P3010 | ALL | 2 | M | 11.0 | Flu/Cy/TBI | 5 | CSA, MMF | Positive | 191 | 13.9 |
| P3275 | SCID | 1 | M | 5.2 | Bu/Cy/Flu | 6 | CSA, MMF, steroids | Positive | 121 | 18.8 |
| P3317 | Myelofibrosis | 2 | M | 9.7 | Bu/Cy/Flu | 5 | CSA, MMF, steroids | Positive | 225 | 18.6 |
| P3457 | JMML | 2 | M | 12.3 | Bu/Cy/Flu | 5 | CSA, MMF | Positive | 185 | 12.0 |
| P3531 | SCID | 1 | M | 6.4 | Bu/Cy/Flu | 5 | CSA, MMF, steroids | Positive | 129 | 16.0 |
| P3555 | SCID | 1 | M | 8.0 | Bu/Cy/Flu | 6 | CSA, MMF, steroids | Positive | 110 | 11.0 |
| P3923 | AML | 1 | F | 7.0 | Bu/Cy/Flu | 5 | CSA, MMF, steroids | Positive | 102 | 11.7 |
| P3626 | JMML | 1 | M | 9.0 | Bu/Cy/Flu | 5 | CSA, MMF | Positive | 209 | 18.6 |
| ACT-CAT2 | ||||||||||
| P0020 | AML | 3 | F | 16.7 | Bu/Cy | 5 | CSA, MMF, steroids, ECP | Positive | 103 | 4.9 |
| P0035 | Hurler’s syndrome | 1.1 | F | 8.8 | Bu/Flu/ATG/ rituximab | 4 | CSA, steroids | Positive | 252 | 22.9 |
| P0039 | Infant ALL | 1.9 | M | 12.2 | Flu/Cy/TBI | 6 | CSA, MMF, steroids | Positive | 67 | 4.4 |
| P0067 | Sickle cell disease | 8 | F | 19.8 | Flu/Mel/Thio/alemtuzumab/HU | 6 | CSA, MMF, infliximab, steroids | Positive | 109 | 4.4 |
| P0123 | Sickle cell disease | 4 | M | 16.6 | Flu/Mel/Thio/alemtuzumab/HU | 5 | CSA, tacro, MMF, steroids | Negative | 197 | 9.5 |
| Patient ID | Diagnosis | Age, y | Sex | Weight, kg | Conditioning regimen | HLA match (of 6) | Immunosuppression before VST | Recipient CMV serostatus | TNCs from UCB, × 107 | TNCs/kg of UCB fraction used for primary transplant |
|---|---|---|---|---|---|---|---|---|---|---|
| ACT-CAT | ||||||||||
| P2891 | Fanconi anemia | 5 | M | 13.0 | Flu/Cy/TBI | 5 | CSA,MMF, steroids | Positive | 161 | 9.9 |
| P3010 | ALL | 2 | M | 11.0 | Flu/Cy/TBI | 5 | CSA, MMF | Positive | 191 | 13.9 |
| P3275 | SCID | 1 | M | 5.2 | Bu/Cy/Flu | 6 | CSA, MMF, steroids | Positive | 121 | 18.8 |
| P3317 | Myelofibrosis | 2 | M | 9.7 | Bu/Cy/Flu | 5 | CSA, MMF, steroids | Positive | 225 | 18.6 |
| P3457 | JMML | 2 | M | 12.3 | Bu/Cy/Flu | 5 | CSA, MMF | Positive | 185 | 12.0 |
| P3531 | SCID | 1 | M | 6.4 | Bu/Cy/Flu | 5 | CSA, MMF, steroids | Positive | 129 | 16.0 |
| P3555 | SCID | 1 | M | 8.0 | Bu/Cy/Flu | 6 | CSA, MMF, steroids | Positive | 110 | 11.0 |
| P3923 | AML | 1 | F | 7.0 | Bu/Cy/Flu | 5 | CSA, MMF, steroids | Positive | 102 | 11.7 |
| P3626 | JMML | 1 | M | 9.0 | Bu/Cy/Flu | 5 | CSA, MMF | Positive | 209 | 18.6 |
| ACT-CAT2 | ||||||||||
| P0020 | AML | 3 | F | 16.7 | Bu/Cy | 5 | CSA, MMF, steroids, ECP | Positive | 103 | 4.9 |
| P0035 | Hurler’s syndrome | 1.1 | F | 8.8 | Bu/Flu/ATG/ rituximab | 4 | CSA, steroids | Positive | 252 | 22.9 |
| P0039 | Infant ALL | 1.9 | M | 12.2 | Flu/Cy/TBI | 6 | CSA, MMF, steroids | Positive | 67 | 4.4 |
| P0067 | Sickle cell disease | 8 | F | 19.8 | Flu/Mel/Thio/alemtuzumab/HU | 6 | CSA, MMF, infliximab, steroids | Positive | 109 | 4.4 |
| P0123 | Sickle cell disease | 4 | M | 16.6 | Flu/Mel/Thio/alemtuzumab/HU | 5 | CSA, tacro, MMF, steroids | Negative | 197 | 9.5 |
ALL, acute lymphocytic leukemia; AML, acute myeloid leukemia; ATG, anti-thymocyte globulin; Bu, busulfan; CSA, cyclosporin A; Cy, cyclophosphamide; ECP, extracorporeal photopheresis; F, female; Flu, fludarabine; HLA, human leukocyte antigen; HU, hydroxyurea; JMML, juvenile myelomonocytic leukemia; M, male; Mel, melphalan; MMF, mycophenolate mofetil; SCID, severe combined immunodeficiency; tacro, tacrolimus; TBI, total body irradiation; Thio, thiotepa; TNC, total nucleated cells; UCB, umbilical cord blood.
Feasibility of manufacturing CB-VSTs
Of 21 patients who had VST manufacture initiated on the protocols, 18 patients had VSTs that were successfully manufactured for clinical use (supplemental Table 1). On ACT-CAT2, manufacturing for 2 VSTs failed to meet lot release because of insufficient cell recovery, and there was 1 potential contamination during separation of the 20% fraction. Fourteen of these 18 patients with successfully generated products were infused with CB-VSTs. Reasons for not infusing were GVHD (n = 2; 1 on ACT-CAT and 1 on ACT-CAT2), death (n = 1; ACT-CAT2), and family choice (n = 1; ACT-CAT2). Overall, the median manufacturing time (from initiation to clinical freeze and excluding release testing) was 52.5 days (range, 40-111 days). Delays in manufacturing were a result of either slow growth of EBV LCLs or the requirement to restimulate cryopreserved products. Including time required for release testing and other variables, the median time to infusion was 87 days (range, 63-434 days).
Engraftment
The primary transplant used 80% of the CB unit (Figure 1). At a minimum cell dose for primary transplant of 2.5 × 107 TNCs/kg,34 all patients engrafted within expected time frames35 with a median time to neutrophil engraftment of 18 days (range, 11-28 days) and a median time to platelet engraftment of 51 days (range, 35-66 days).
Product characterization
CB-VSTs were expanded by using APCs transduced with an Ad5F35-pp65 vector or with APCs pulsed with overlapping peptides (PepMixes) representing the same viruses. After 3 expansions, the median number of the CB-VSTs manufactured from each 20% fraction was 6 × 107 cells (range, 3.3 to 27.7 × 107 cells) with higher cell yields in the peptide-based manufacture (ACT-CAT2) compared with viral vector–based manufacture (ACT-CAT) (143 × 107 vs 43 × 107 cells; P = .001) (Figure 2A). Most VSTs were specific for more than 1 virus (Figure 2B), with a median SFC of 13 (range, 1-110 SFCs) for CMV-pp65, 19 for Ad-hexon (range, 0-486 SFCs), and 42 for EBV LCLs or EBV-EBNA1/LMP2 (range, 0-339 SFCs). For products made under ACT-CAT2, PepMixes for EBV-EBNA1, EBV LMP2, and CMV-IE1 were also evaluated when possible alongside other PepMixes and EBV LCLs. Expanded cells were composed of a median of 50% CD3+/CD4+ (range, 5% to 86% CD3+/CD4+) and 32% CD3+/CD8+ (range, 4% to 79% CD3+/CD8+) T cells with negligible CD19+ and CD3–/CD83+ APCs. Absence of these defined APCs was a release criterion for both protocols. CB-VSTs composed a median of 39% (range, 22% to 69%) effector memory (CD45RA–/CD62L–) and a median of 38% (range, 16% to 74%) central memory (CD45RA–/CD62L+) T cells (Figure 2C). The CD45RA+ population was too small to fully characterize.
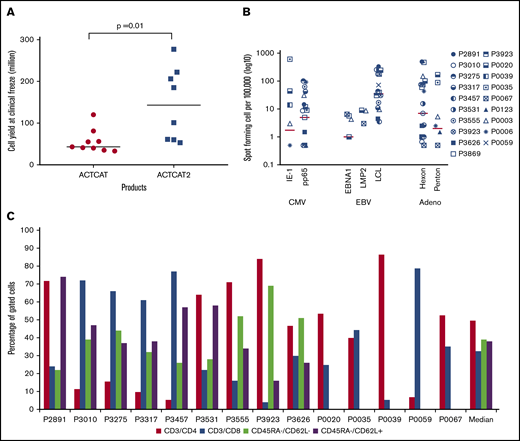
VST product characteristics. (A) Cell yields after manufacture using APCs either transduced with the Ad5f35-pp65 vector (ACT-CAT) or cocultured with overlapping viral peptides (ACT-CAT2). (B) VST products demonstrated multivirus specificity as measured by IFN-γ ELISPOT assay in response to CMV, EBV, and adenovirus antigen. Data for 19 VST products is represented as the number of SFCs per 100 000 after subtracting the values of the actin negative control. (C) A CD4+ T-cell predominant phenotype was observed with similar effector memory (CD45RA–/CD62L–) and central memory (CD45RA–/CD62L+) percentages in the VST products. Memory phenotype data were not available for products P0020, P0035, P0039, P005, and P0067 because of limited product availability for analysis after infusion.
VST product characteristics. (A) Cell yields after manufacture using APCs either transduced with the Ad5f35-pp65 vector (ACT-CAT) or cocultured with overlapping viral peptides (ACT-CAT2). (B) VST products demonstrated multivirus specificity as measured by IFN-γ ELISPOT assay in response to CMV, EBV, and adenovirus antigen. Data for 19 VST products is represented as the number of SFCs per 100 000 after subtracting the values of the actin negative control. (C) A CD4+ T-cell predominant phenotype was observed with similar effector memory (CD45RA–/CD62L–) and central memory (CD45RA–/CD62L+) percentages in the VST products. Memory phenotype data were not available for products P0020, P0035, P0039, P005, and P0067 because of limited product availability for analysis after infusion.
Safety of CB-VSTs
The ACT-CAT and ACT-CAT2 trials were designed to evaluate 4 dose levels: 5 × 106, 1.0 × 107, 1.5 × 107, and 2.5 × 107 cells per m2. In ACT-CAT, no treatment-related DLT was observed, and the study dose was escalated as planned after infusion of 2 patients at each of the first 3 dose levels (5 × 106, 1.0 × 107, and 1.5 × 107 cells per m2) and concluded after 3 patients were treated at the highest dose level of 2.5 × 107 cells per m2 without a DLT. In ACT-CAT2, 4 patients were treated at the first dose level (5 × 106 cells per m2) followed by escalation to the second dose level at which patients received 1 × 107 cells per m2, and again no treatment-related DLT was observed. No infusion-related adverse events were observed in any of the 14 patients treated on either trial. No patient developed grade 3 to 4 acute GVHD during the 45-day post infusion monitoring period. One patient (P0067) with improving GVHD at the time of VST infusion developed grade 3 acute GVHD involving the gastrointestinal tract 53 days after VST; within 4 days of treatment with systemic steroids, the patient experienced complete resolution of symptoms and was subsequently weaned off all immunosuppression without further recurrence.
Viral infections
VSTs were infused at a median of 87 days (range, 48-434 days) after transplantation (supplemental Table 1). Thirteen of 14 patients were receiving immunosuppression with a combination of calcineurin inhibitors, mycophenolate mofetil, or low-dose steroids at the time of infusion. One patient in the treatment group (P2891) received 3 VST infusions for persistent disease, and 1 in the prophylaxis group (P0020) received 2 infusions for viral reactivation after the first infusion (Table 2). Of the 14 patients treated (Table 2), 7 had detectable low-level viral reactivation or disease within 1 week before CB-VST infusion. The remaining 7 were deemed to be at high risk for developing viral disease and received CB-VST prophylactically. All 7 patients with detectable virus had a decrease in viral burden after CB-VST infusion. Five patients cleared the virus at a median of 2 months (range, 1.4-12 months) after CB-VST. Patient P0067 cleared CMV from the blood but received steroid therapy after VST infusion for GVHD. This patient was subsequently diagnosed with CMV retinitis 6 months after CB-VST infusion and was taken off study to receive third-party VSTs because the steroid therapy for GVHD likely affected CB-VST persistence in vivo. Patient P3457 continued to have low levels of EBV DNA but did not develop posttransplant lymphoproliferative disorder and did not receive additional treatment (eg, anti-CD20 therapy). Of the 7 patients at high risk for viral infection who received the CB-VSTs as prophylaxis, no patient developed end organ disease and only 1 patient (P0020) required additional therapy (second CB-VST infusion and cidofovir) to treat adenovirus reactivation in the blood and stool.
Patient infection/reactivation status and outcome
| Patient | Indication | Prior therapy | Viral course after VST | Status within first year |
|---|---|---|---|---|
| P0123 | Adenovirus low-level viremia (<200 copies per mL) and positive upper respiratory tract | Cidofovir | Resolution (with brincidofovir) | Alive, GVHD resolved at 1 y |
| P0067 | CMV 2239 cells per mL | Foscarnet/cidofovir/ganciclovir | CMV viremia resolved by 6 mo but developed CMV retinitis 6 mo after infusion | Alive, CMV retinitis at 6 mo treated with third-party T cells and intravitreal ganciclovir with complete resolution, GVHD resolved at 1 y |
| P0039 | CMV low-level viremia (<1000 copies per mL) | Foscarnet/cidofovir/ganciclovir | Resolution | Leukemia relapse after 90 d, in remission after CAR T-cell therapy |
| P0035 | CMV low-level viremia (<1000 copies/mL) and positive stool | Foscarnet | Resolution (with valganciclovir) | Alive, quiescent GVHD on cyclosporine at 1 y |
| P2891 | CMV viremia (100 copies/mL) | Foscarnet, CMV immunoglobulin | Resolution (with ganciclovir + 2 additional CB-VST infusions) | Alive, GVHD resolved at 1 y |
| P3626 | EBV in bone marrow (3500 copies/mL) | Cidofovir for previous adenoviremia | Fivefold decline in bone marrow EBV at 5 mo, negative in blood at 1 y, no PTLD | Alive, MDS + monosomy 7 at <1 y resolved at 2 y. No GVHD, off immunosuppression at 1 y |
| P3457 | EBV viremia (109 copies/mL) | None | Persistent viremia but no PTLD | Died after second transplant for relapsed leukemia |
| P3010 | Prophylaxis | Ganciclovir prophylaxis | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P3275 | Prophylaxis | None | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P3317 | Prophylaxis | Ganciclovir prophylaxis | No reactivation requiring therapy, no disease | Relapse/AML progression <1 y after HCT, deceased day +284. |
| P3531 | Prophylaxis | Valganciclovir prophylaxis | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P3555 | Prophylaxis | None | No reactivation requiring therapy, no disease | Alive, resolved GVHD at 1 y |
| P3923 | Prophylaxis | None | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P0020 | Prophylaxis/past history of adenovirus in stool | Cidofovir | Adenovirus reactivation in stool at 3 wk and then blood 3 mo after VSTs; given second CB-VST infusion and continued with cidofovir | Alive, quiescent GVHD on cyclosporine at 1 y |
| Patient | Indication | Prior therapy | Viral course after VST | Status within first year |
|---|---|---|---|---|
| P0123 | Adenovirus low-level viremia (<200 copies per mL) and positive upper respiratory tract | Cidofovir | Resolution (with brincidofovir) | Alive, GVHD resolved at 1 y |
| P0067 | CMV 2239 cells per mL | Foscarnet/cidofovir/ganciclovir | CMV viremia resolved by 6 mo but developed CMV retinitis 6 mo after infusion | Alive, CMV retinitis at 6 mo treated with third-party T cells and intravitreal ganciclovir with complete resolution, GVHD resolved at 1 y |
| P0039 | CMV low-level viremia (<1000 copies per mL) | Foscarnet/cidofovir/ganciclovir | Resolution | Leukemia relapse after 90 d, in remission after CAR T-cell therapy |
| P0035 | CMV low-level viremia (<1000 copies/mL) and positive stool | Foscarnet | Resolution (with valganciclovir) | Alive, quiescent GVHD on cyclosporine at 1 y |
| P2891 | CMV viremia (100 copies/mL) | Foscarnet, CMV immunoglobulin | Resolution (with ganciclovir + 2 additional CB-VST infusions) | Alive, GVHD resolved at 1 y |
| P3626 | EBV in bone marrow (3500 copies/mL) | Cidofovir for previous adenoviremia | Fivefold decline in bone marrow EBV at 5 mo, negative in blood at 1 y, no PTLD | Alive, MDS + monosomy 7 at <1 y resolved at 2 y. No GVHD, off immunosuppression at 1 y |
| P3457 | EBV viremia (109 copies/mL) | None | Persistent viremia but no PTLD | Died after second transplant for relapsed leukemia |
| P3010 | Prophylaxis | Ganciclovir prophylaxis | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P3275 | Prophylaxis | None | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P3317 | Prophylaxis | Ganciclovir prophylaxis | No reactivation requiring therapy, no disease | Relapse/AML progression <1 y after HCT, deceased day +284. |
| P3531 | Prophylaxis | Valganciclovir prophylaxis | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P3555 | Prophylaxis | None | No reactivation requiring therapy, no disease | Alive, resolved GVHD at 1 y |
| P3923 | Prophylaxis | None | No reactivation requiring therapy, no disease | Alive, no GVHD, off immunosuppression at 1 y |
| P0020 | Prophylaxis/past history of adenovirus in stool | Cidofovir | Adenovirus reactivation in stool at 3 wk and then blood 3 mo after VSTs; given second CB-VST infusion and continued with cidofovir | Alive, quiescent GVHD on cyclosporine at 1 y |
CAR, chimeric antigen receptor; HCT, hematopoietic stem cell transplantation; MDS, myelodysplastic syndrome; PTLD, posttransplantation lymphoproliferative disorder.
Virus-specific immune reconstitution was superior in patients receiving CB-VSTs at the time of viral infection or reactivation
VST responses were measured at the time of infusion and periodically thereafter by interferon-γ ELISPOT assay in response to stimulation with viral antigens (Figure 3A). Figure 3A shows data from a representative patient (P0039) with CMV reactivation in which the viral load increases and then clears after an increase in pp65-specific T cells in the patient’s peripheral blood. Patients who were treated for active viral infections (n = 7) (Figure 3B) seemed to have superior VST immune reconstitution at 3 months after infusion compared with those patients treated prophylactically without viral infection or reactivation (n = 7) (Figure 3C).
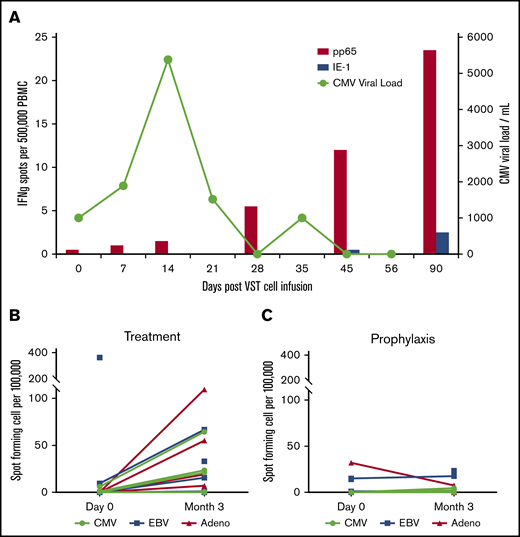
T-cell persistence and viral load in patients treated with CB-VSTs. (A) Detection of CMV-specific T cells in the peripheral blood of patient P0039 (red bars) and the CMV viral load (green line). Detection of CMV-, EBV-, and adenovirus-specific T cells at baseline compared with 3 months after VST infusion in the viral treatment group (B) and the prophylaxis group (C).
T-cell persistence and viral load in patients treated with CB-VSTs. (A) Detection of CMV-specific T cells in the peripheral blood of patient P0039 (red bars) and the CMV viral load (green line). Detection of CMV-, EBV-, and adenovirus-specific T cells at baseline compared with 3 months after VST infusion in the viral treatment group (B) and the prophylaxis group (C).
Long-term persistence of CB-VSTs as detected by using TCR sequencing
In the absence of gene marking, tracking infused CB-VSTs in patients is difficult. However, by sequencing TCR clonotypes present in the CB-VST products, VSTs can be tracked in the peripheral blood over time on the basis of the presence of clonotypes associated with the CB-VST product. Figure 4A-C shows unique clonotypes present in the manufactured VST product and detected in the patients’ peripheral blood after VST infusion but not detected in the patients’ peripheral blood before infusion. As demonstrated in 1 patient (P2891) with viral infection at the time of infusion, there was an increase in the frequency of unique clonotypes after infusion of CB-VSTs (Figure 4A). This was also observed in 2 patients (P3555 and P3317) with no viral infection at the time of infusion (Figure 4B-C). We also observed an increase in the naïve T-cell (CD45RA+) TCR-Vβ diversity over time in 1 patient (P2891) who had CMV viremia at the time of CB-VST infusion, further supporting a restoration of immunity with infusion (Figure 4D). The persistence of TCR clonotypes unique to the CB-VST product was evident up to 1 year after infusion (Figure 4H), and these unique clones persisted regardless of the presence of viral antigen at the time of infusion (Figure 4I).
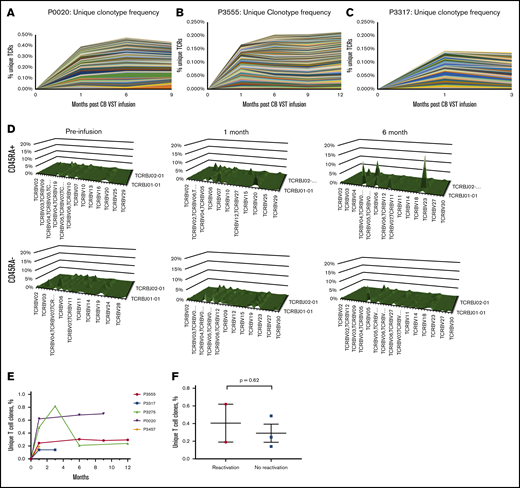
Detection and persistence of unique TCRs over time using TCR sequencing. (A-C) Shown are unique clonotypes that were present in the VST cell product but not present before VST cell infusion. (D) TCR-Vβ diversity over time in patient P2891 in CD45RA+ (naïve) and CD45RA– (memory) T-cell populations. (E) Persistence of unique T-cell clones over time in all patients with available data. (F) Unique T-cell clone contribution at 1 month after VST infusion separated by whether patients had detectable virus within the first month of infusion (reactivation) or not (no reactivation).
Detection and persistence of unique TCRs over time using TCR sequencing. (A-C) Shown are unique clonotypes that were present in the VST cell product but not present before VST cell infusion. (D) TCR-Vβ diversity over time in patient P2891 in CD45RA+ (naïve) and CD45RA– (memory) T-cell populations. (E) Persistence of unique T-cell clones over time in all patients with available data. (F) Unique T-cell clone contribution at 1 month after VST infusion separated by whether patients had detectable virus within the first month of infusion (reactivation) or not (no reactivation).
Discussion
Here we evaluated the use of CB-VSTs generated from the 20% fraction of CB units used for CBT. Small product volume, low cell numbers, high percentage of nucleated red blood cells, and inability to procure further donations have prevented the widespread use of CB cells as a source of cellular therapy beyond transplantation. Whereas chimeric antigen receptor T cells,36 VSTs,37-39 and other cellular therapies generated from donor PBMCs have recently demonstrated success, to our knowledge, this is the first clinical study using 20% of a fractionated CB unit intended for transplant as a means to augment the graft. In this study, we demonstrated that using the 20% fraction is feasible, results in sufficient expansion of cells to treat patients on clinical protocols, and may provide clinical benefit to patients.
Recently, many groups have confirmed the efficacy of antiviral T cells from seropositive donors. Such antiviral T cells can now be manufactured in 10 days40 with products including VSTs that target up to 6 viruses41 in a single culture. By using these approaches, the overall response rate has exceeded 90%.38 For more rapid cultures, Peggs and colleagues42 showed that it was feasible to generate sorted VST products in less than 48 hours that were safe and efficacious. However, all of these reports required the donor to be seropositive for each of the viruses targeted. In this report, we have used CB as the T-cell source, which contains predominantly virus-naïve T cells. Because the T cells are derived from the naïve T-cell population, the manufacture requires additional cytokines, stimulation with professional APCs, and a longer manufacturing time.
The use of naïve T cells as a source of VSTs has not been extensively studied, although murine studies suggest that naïve-derived antigen-specific T cells have superior proliferation, longer telomeres, and better function than certain memory-derived T cells.26,27 Although this may be true in vivo, it is important to note that the ex vivo expansion of T cells can be difficult, especially given the limited starting volume and cell number. Nevertheless, the required cell dose was met for all but 2 patients, in whom there were abnormally low cell counts before initiating the culture. Without a blinded, head-to-head trial comparing memory-derived vs naïve-derived VSTs, it is impossible to determine whether the naïve-derived T cells are as efficacious. The complexity of the patients receiving VSTs and the small sample size also make assessing the efficacy of the T cells difficult. Nevertheless, the ability to generate donor-derived CB-VSTs that can be administered as prophylaxis to high-risk patients after CBT to prevent viral disease and restore antiviral immunity long term is an attractive approach. Other challenges still remain, such as generating sufficient expansion to yield adequate cells for large adult recipients. On the basis of the highest dose level tested in this study of 2.5 × 107 cells per m2 and a theoretical body surface area of 2 m2, 100% of patients would have been eligible to receive CB-VSTs using the optimized manufacturing on the second trial. With further refinements to the manufacturing process such as those previously reported as well as a higher desired manufacturing yield, it is possible that the percentage of eligible recipients would be higher, although additional optimization is needed to treat large adult patients. The feasibility of this will be assessed in our follow-up study (ClinicalTrials.gov identifier: NCT03594981).31
Previous work by our group indicated that naïve-derived T cells recognize atypical epitopes of CMV.12 The CB-VSTs manufactured for our 2 clinical trials were expected to elicit T-cell responses in vivo that recognize only atypical epitopes of CMV-pp65. However, the limited volume of blood obtained from the patients for follow-up testing and the lack of CMV reactivation or infection in patients for which epitope characterization was available made evaluating the persistence of these unique T-cell populations challenging. We plan to perform these analyses in future clinical trials that treat adult patients with CB-VSTs in which follow-up patient samples should be adequate. Regardless, we did observe the persistence of CMV-specific T-cell clones by deep TCR sequencing.28
In this study, we demonstrated the feasibility of VST manufacture from the 20% fraction of naïve CB in the majority of patients enrolled on the study. However, 3 of 21 products failed manufacturing, and the overall time-to-clinical freeze of 52.5 days remains an obstacle to widespread adoption of this treatment. Current approaches for antiviral adoptive cellular therapy require less than 10 days for ex vivo expansion, with cell selection approaches requiring less than 2 days. Improvements have been made to this process, and a 30-day manufacturing method has been reported,31 but it remains to be seen whether the time can be reduced to 10 to 14 days and therefore be more readily applicable worldwide instead of limited to specialized individual centers. In addition, because the CB units are cryopreserved, the VST products could be manufactured before transplantation and be available when needed.
In summary, we have demonstrated the safety and feasibility of using the 20% fraction of fractionated CB units to expand CB-VSTs for clinical use. When administered to patients after CBT, CB-VSTs may persist over the long term and restore antiviral immunity to prevent viral infection or may complement antiviral pharmacotherapies when administered for the treatment of active CMV, EBV, or adenovirus infection in high-risk patients.
Acknowledgments
The authors thank Helen Heslop for her mentorship and assistance with the study and April Durrett (Baylor College of Medicine) for her flow cytometry expertise.
This work was supported by a Program Project grant from the National Institutes of Health, National Cancer Institute (P01 CA148600) (E.J.S. and C.M.B.), the Amy Strelzer Manazevit Program (P.J.H.), the American Society of Hematology/Robert Wood Johnson Harold Amos Medical faculty development program (A.A.A.), and the American Cancer Society (PF-13e046e01-LIB) (P.J.H.).
Authorship
Contribution: C.M.B., P.J.H., H.L., and E.J.S. conceived and designed the study; C.A.M., A.A.A., A.P.G., P.J.H., L.R., D.A.J., and R.A.K. provided study materials or patients; A.A.A., P.J.H., T.D.J., M.D.K., B.S., H.D., C.R.Y.C., B.J.G., F.H., and C.M.B. collected and assembled the data; A.A.A., P.J.H., H.L., T.D.J., C.A.M., and C.M.B. analyzed and interpreted the data; and all authors wrote the manuscript, provided final approval, and were accountable for all aspects of the work.
Conflict-of-interest disclosure: C.M.B. is on the scientific advisory board (SAB) for Cellectis, has stock options in Neximmune and Torque Therapeutics, and is a cofounder and SAB member of Mana Therapeutics. P.J.H., C.R.Y.C., M.D.K., and C.M.B. have patents related to this work. P.J.H. and C.R.Y.C. are cofounders of Mana Therapeutics and serve on the Board (P.J.H.) or the SAB (C.R.Y.C.). The remaining authors declare no competing financial interests.
Correspondence: Catherine M. Bollard, Center for Cancer and Immunology Research, Children’s National Health System, 111 Michigan Ave NW, Washington, DC 20010; e-mail: cbollard@childrensnational.org.

