

Key Points
GSK3β is upregulated and more nuclear in localization in AML, allowing it to promote a more aggressive disease in vivo and in vitro.
Nuclear localization of GSK3β is correlated with poorer overall survival and drug resistance.

Abstract
Acute myeloid leukemia (AML) is a devastating disease with poor patient survival. As targetable mutations in AML are rare, novel oncogenic mechanisms are needed to define new therapeutic targets. We identified AML cells that exhibit an aberrant pool of nuclear glycogen synthase kinase 3β (GSK3β). This nuclear fraction drives AML growth and drug resistance. Nuclear, but not cytoplasmic, GSK3β enhances AML colony formation and AML growth in mouse models. Nuclear GSK3β drives AML partially by promoting nuclear localization of the NF-κB subunit, p65. Finally, nuclear GSK3β localization has clinical significance as it strongly correlates to worse patient survival (n = 86; hazard ratio = 2.2; P < .01) and mediates drug resistance in cell and animal models. Nuclear localization of GSK3β may define a novel oncogenic mechanism in AML and represent a new therapeutic target.
Introduction
Acute myeloid leukemia (AML) is an aggressive disease with poor patient survival. Although a wide range of genetic abnormalities has been described in AML, individual patients exhibit only a small number of these abnormalities, and these vary considerably among patients.1,2 This molecular heterogeneity complicates the development of targeted AML therapeutics with broad efficacy. Identifying more universal abnormalities in AML would provide novel targets and improved prognostic biomarkers.
Glycogen synthase kinase 3 (GSK3) has been reported as a promising AML therapeutic target.3-5 GSK3 is a kinase that affects multiple signaling pathways important for cellular self-renewal, growth, and survival, including the NF-κB and β-catenin pathways, both critical for AML development.6,7 GSK3 inhibition promotes growth inhibition and differentiation of a variety of AML cells, irrespective of specific mutations.3,5 Unlike traditional AML chemotherapy that exhibits toxic effects on normal hematopoietic cells as well as AML cells, for unknown reasons GSK3 inhibition does not lead to growth impairment of normal hematopoietic cells.3-5 In fact the GSK3 inhibitor, lithium, has been used clinically to accelerate the recovery of white blood cells after bone marrow transplantation.8
GSK3 inhibitors target both isoforms of GSK3, GSK3α and GSK3β, and we and others have shown GSK3β plays an important role in AML.3,5 GSK3β upregulation has been observed in a wide variety of cancers, including pancreatic, non–small cell lung, gastric, and chronic lymphocytic leukemia.9-15 Interestingly, in chronic lymphocytic leukemia and pancreatic cancer, GSK3β aberrantly localizes to the nucleus. This nuclear localization of GSK3β has been correlated to the expression of antiapoptotic genes such as Bcl-2 and Bcl-XL.11,16 Although nuclear GSK3β has been observed in several types of cancer, whether the upregulation of nuclear GSK3β specifically could promote more aggressive cancer is less clear.
Here we show GSK3β protein is universally upregulated and aberrantly nuclear localized in AML as compared with normal hematopoietic cells. Then utilizing a targeted, inducible GSK3β rescue system, we show increased nuclear localized GSK3β, not merely increased GSK3b expression, promotes more aggressive, drug-resistant AML. Finally, we show that AML patient samples exhibit a wide variation in nuclear GSK3β partitioning, and that patients with the most pronounced nuclear GSK3β localization exhibit poorer survival.
Materials and methods
Reagents and cells
Daunorubicin and doxycycline were from Dot Scientific (Burton, MI). 5-Fluorouracil was from TCI America (Portland, OR). JSH-23 was obtained from Apex Biotechnology Corp. (Taiwan). Antibodies were from the following: hemagglutinin (HA)-tag, caspase-3, poly ADP ribose polymerase (PARP), GSK3β, pGSK3β, p65, pp65 (S536), IκB, glyceraldehyde-3-phosphate dehydrogenase, Histone 3, X-linked inhibitor of apoptosis protein (XIAP), and Bcl-XL (Cell Signaling Technologies, Beverly MA); CD34 (Beckman Coulter, Brea, CA); CD38, CD14, and CD15 (BD Biosciences, San Jose, CA); and CD3, CD117, and CD19 (Biolegend). P65-A488 was obtained from Santa Cruz Biotechnology (Dallas, TX). Quantitative polymerase chain reaction (qPCR) primers were obtained from Sigma-Aldrich (GSK3β: F 5′-GGAACTCCAACAAGGGAGCA-3′, R 5′-TTCGGGGTCGGAAGACCTTA-3′; β-actin: F 5′-AGAGCTACGAGCTGCCTGAC-3′, R 5′-AGCACTGTGTTGGCGTACAG-3′). Cell lines were from the following: OCI-AML3 (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany); and 293T, Phoenix, U937, THP-1, and HL60 cells (American Type Culture Collection, Manassas, VA). Primary AML and normal donor samples were from the Case Western Reserve University Hematopoietic Cell Core Facility (Cleveland, OH) or Eastern Cooperative Oncology Group–American College of Radiology Imaging Network (ECOG-ACRIN) E1900 and E3999. Mouse progenitor cells were obtained as previously described.17
GSK3β rescue system
To establish targeted GSK3β constructs, GSK3β was mutated to S9A. Three silent mutations (C1847T, A1850T, and A1853G) were made using the Quickchange Lightning kit (Agilent Technologies, Santa Clara, CA). For GSK3β–nuclear localization signal (NLS), 3 SV40 large T NLS were added to the GSK3β C terminus. GSK3β-Cyto was made as previously described, mutating R102G and K103A.18 GSK3β complementary DNA (cDNA) was cloned into PLVX-TRE3G-EF1a-mCherry vector from Clontech (Mountain View, CA). Each construct was HA-tagged at the N terminus.
Cells were transduced with GSK3β shRNA TRCN0000039999 (Sigma, St. Louis, MO) and selected with blasticidin. Next, GSK3β-Cyto or GSK3β-NLS and a tet-regulator were transduced (Tet-on 3G, Clontech) and selected using puromycin and G418. GSK3β expression was induced for a minimum of 48 hours with doxycycline (0.2-1.5 μg/mL).
qPCR
Total RNA was isolated from either AML cells lines or peripheral blood mononuclear cells (PBMCs) from healthy donors and AML patients using the RNeasy Mini Kit (Qiagen, Hilden, Germany). RNA was transcribed into cDNA using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA). qPCR was performed in quadruplicates using EvaGreen Dye (Biotium, Fremont, CA) on a BIO-RAD CFX96 Real-Time Thermocycler (Bio-Rad, Hercules, CA).
Viral transductions
Lentiviral.
293T and AML cells were transfected as previously described.3
Retroviral.
Phoenix E cells were cotransfected with MLL-AF9-GFP (Addgene) or vector and PCL7 (Addgene). After 48 hours, cells were concentrated with Retro-X concentrator per the manufacturer’s instructions (Takara Bio, Japan). Cells were incubated in viral supernatant in retronectin-coated plates for 3 days (Takara Bio).
Western blot
Cells treated as indicated were lysed with a triton containing lysis buffer for whole cell extracts. Western blots were performed as previously described.3,19
Mouse xenograft studies
Six-week-old female Nod/SCID/IL2-Rγ−/− (NSG) mice (Jackson Laboratory, Bar Harbor, ME) were injected IV with 3 million OCI-AML or HL60 cells per group (n = 5 per group). GSK3β expression was induced by administering 100 μg/mL doxycycline ad libertum. In the drug resistance study, 4 days after cell injection, daunorubicin was administered once daily to half of the induced and control mice at a dose of 1.5 mg/kg for a 3 doses. Mice were euthanized according to our institutional guidelines (signs of significant disease morbidity such as limb paralysis or >20% weight loss).
Colony assay
Ten thousand viable cells per group were plated in 1.27% methylcelluose (R&D Systems, Minneapolis, MN). Colonies were counted after 7 to 10 days.
Drug resistance assays
Viability was assessed by counting at least 100 cells in at least 2 representative microscope fields using trypan blue (Thermo Fisher) or Hoechst 33342 staining (Invitrogen, Carlsbad, CA).
Luciferase assay
293T cells were transfected with a GSK3β construct, ELAM-luciferase, and Renilla-Luciferase construct. After 48 hours, cells were analyzed using the Biotium Dual Firefly/Renilla Luciferase Kit per manufacturer instructions (Fremont, CA).
Flow cytometry and imaging cytometry
Cell surface markers were stained for 15 minutes at room temperature (CD34 or CD117 for ECOG samples). Cells were then fixed in 4% formaldehyde and stained per antibody supplier instruction but with 80% methanol and analyzed on a LSR II (BD Biosciences) or Attune NXT cytometer (ThermoFisher) or an Amnis ImageStream cytometer for imaging cytometry (EMD Millipore Billerica, MA). Before each run, BD calibrite beads were run to normalize for day-to-day variation. The nuclear localization score was calculated using IDEAS 6.1 software (EMD Millipore).
Statistical analysis
The Student t test and Wilcoxon rank-sum test were used to compare the distribution of continuous values in 2 groups. Error bars are defined as ±1 standard error. Mouse survival data were analyzed using the log-rank test. Multiple means was analyzed using the Tukey Kramer test or 1-way analysis of variance (not significant, P > .05; *P < .05; **P < .01; ***P < .001). Cook’s distance was used to determine regression model outliers. The biomarker studies were performed in a blinded fashion between laboratory investigators and a statistician. Survival distributions were estimated using the Kaplan-Meier method. Cox proportional hazards models were used to assess the association between overall survival and covariates.
Study approval
The Case Western Reserve University Institutional Animal Care and Use Committee approved all of the animal protocols used in this study. For the human patient sample work, written informed consent was received from participants prior to inclusion in the study. The University Hospitals Cleveland Medical Center Institutional Review Board approved all of the human subject’s research.
Results
GSK3β is upregulated and exhibits aberrant nuclear localization in AML
GSK3 inhibition and knockdown selectively leads to the growth inhibition and differentiation of AML cells while sparing normal hematopoietic cells, suggesting GSK3 may promote AML maintenance and progression.3-5 To explain this marked difference in biological effects, we probed the protein expression and subcellular localization of GSK3β in AML and normal hematopoietic cells.
Western blot analysis comparing normal bone marrow cells (abbreviated NM), immature peripheral blood granulocyte colony-stimulating factor mobilized leukocytes (abbreviated MB), and a variety of AML cell lines and primary patient samples showed a dramatic increase in GSK3β protein in AML as compared with normal cells (Figure 1A; supplemental Figure 1). Flow cytometry revealed GSK3β protein levels in normal hematopoietic progenitor cells (CD34+), monocytes (CD14+), B cells (CD19+), neutrophils (CD15+), and T cells (CD3+) were all 5- to 10-fold lower than GSK3β levels in AML (Figure 1B; n = 2 for each subset tested). Finally, GSK3β expression in adult AML diagnosis samples (n = 86) from ECOG-ACRIN studies (E3999 and E1900) is markedly upregulated compared with normal CD34+ hematopoietic progenitor cells (n = 12), suggesting GSK3β elevation is broadly characteristic of AML (P < .001) (Figure 1C-E).
![GSK3β is highly upregulated, active, and more nuclear in AML cells. (A) GSK3β levels were assessed as indicated in normal bone marrow (NM), immature granulocyte colony-stimulating factor mobilized peripheral blood leukocytes (MB), and AML cells by western blot. FAB subtypes are as follows: HL60-M2 OCI-M4 U937-M4 subtype THP1-M5 subtype Pt1-M4 Pt2-M2 Pt3-M4EO. (B) GSK3β levels in different populations of normal cells (n = 2 for each subpopulation) and AML cells (n = 2 for 3 cell lines and 1 patient sample) were analyzed using flow cytometry analysis. P < .001 for all comparisons except for OCI/CD3, Pt/CD3, OCI/CD19, Pt/CD19, OCI/CD14, and Pt/CD14 where P < .01. (C) Imaging cytometry was used to quantify GSK3β expression in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions of GSK3β mean fluorescence intensity. (D) Imaging cytometry was used to quantify GSK3β nuclear localization in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions. (E) Representative images of cells from the cell types indicated from imaging cytometry showing GSK3β expression and the nucleus (4′,6-diamidino-2-phenylindole [DAPI] staining). The GSK3β expression level and nuclear localization of each sample is shown. **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/21/10.1182_bloodadvances.2018016006/4/m_advances016006f1-1.png?Expires=1763469067&Signature=cU9ssLE8VV8hi4FkZZDpAMmpY6Ici3Kjqfk7iz3S~O4DQ2j~34LjgXUmW-xYAGTRvbssMxN7m5DFGcS-Vj5-5AuBCxvLFYF90t7IBKjzO4jSF~IjyGZX6vL5-faBL5hx1uXKO9H2qdy4NSd-OMVnLTu9cuhnb6Czn-k9~QfpFZnzeLSER9aomUpyFi0QmoYE1FWjaGz0gipv76SHLUjfRsAWv3Xb64fkT3L6tL7IJR3nPommZsALMIbTieg8sr79XoWGtpIi~CB35r--t2MNNj22u04ecpUM2kZrG7eWHQnadpIpDlOUT2BwXVbhBZG9sXbNf~eM-tl2Rjrk7crs3Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![GSK3β is highly upregulated, active, and more nuclear in AML cells. (A) GSK3β levels were assessed as indicated in normal bone marrow (NM), immature granulocyte colony-stimulating factor mobilized peripheral blood leukocytes (MB), and AML cells by western blot. FAB subtypes are as follows: HL60-M2 OCI-M4 U937-M4 subtype THP1-M5 subtype Pt1-M4 Pt2-M2 Pt3-M4EO. (B) GSK3β levels in different populations of normal cells (n = 2 for each subpopulation) and AML cells (n = 2 for 3 cell lines and 1 patient sample) were analyzed using flow cytometry analysis. P < .001 for all comparisons except for OCI/CD3, Pt/CD3, OCI/CD19, Pt/CD19, OCI/CD14, and Pt/CD14 where P < .01. (C) Imaging cytometry was used to quantify GSK3β expression in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions of GSK3β mean fluorescence intensity. (D) Imaging cytometry was used to quantify GSK3β nuclear localization in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions. (E) Representative images of cells from the cell types indicated from imaging cytometry showing GSK3β expression and the nucleus (4′,6-diamidino-2-phenylindole [DAPI] staining). The GSK3β expression level and nuclear localization of each sample is shown. **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/21/10.1182_bloodadvances.2018016006/4/m_advances016006f1-2.png?Expires=1763469067&Signature=2cmVXi1B5EKGl8MyA07LYLNmxY~13mFzzkRO0E3gR0sO70h-Bz3Mz2b2R-LQnZiz~Mo4sK8IIfJ3o61iQ-hzEgv9UaQq5uShTtmz7WQOcMUrursTI5hq9jue5v~TFOjiZf7YuRbEQ9a~P1mCnANcSmOTjT4syWvPYG00evqHvUkkcTubjQAbFIqhslD2SMEqlHuoQW-RYla0-A9RSv7yqNeWw~Zw-sVa1YWU9nVeJVqPbv-jVLvgEeXfYUO5ru8WIWxm5eJJy6q5xYQbrgUKkwFdqRgOVKag~RoSjveEmyFJSVcv5gXNLmLG7vdX8DnurTojL88mW7Zu8ZGq9qKBAQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
GSK3β is highly upregulated, active, and more nuclear in AML cells. (A) GSK3β levels were assessed as indicated in normal bone marrow (NM), immature granulocyte colony-stimulating factor mobilized peripheral blood leukocytes (MB), and AML cells by western blot. FAB subtypes are as follows: HL60-M2 OCI-M4 U937-M4 subtype THP1-M5 subtype Pt1-M4 Pt2-M2 Pt3-M4EO. (B) GSK3β levels in different populations of normal cells (n = 2 for each subpopulation) and AML cells (n = 2 for 3 cell lines and 1 patient sample) were analyzed using flow cytometry analysis. P < .001 for all comparisons except for OCI/CD3, Pt/CD3, OCI/CD19, Pt/CD19, OCI/CD14, and Pt/CD14 where P < .01. (C) Imaging cytometry was used to quantify GSK3β expression in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions of GSK3β mean fluorescence intensity. (D) Imaging cytometry was used to quantify GSK3β nuclear localization in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions. (E) Representative images of cells from the cell types indicated from imaging cytometry showing GSK3β expression and the nucleus (4′,6-diamidino-2-phenylindole [DAPI] staining). The GSK3β expression level and nuclear localization of each sample is shown. **P < .01; ***P < .001.
GSK3β is highly upregulated, active, and more nuclear in AML cells. (A) GSK3β levels were assessed as indicated in normal bone marrow (NM), immature granulocyte colony-stimulating factor mobilized peripheral blood leukocytes (MB), and AML cells by western blot. FAB subtypes are as follows: HL60-M2 OCI-M4 U937-M4 subtype THP1-M5 subtype Pt1-M4 Pt2-M2 Pt3-M4EO. (B) GSK3β levels in different populations of normal cells (n = 2 for each subpopulation) and AML cells (n = 2 for 3 cell lines and 1 patient sample) were analyzed using flow cytometry analysis. P < .001 for all comparisons except for OCI/CD3, Pt/CD3, OCI/CD19, Pt/CD19, OCI/CD14, and Pt/CD14 where P < .01. (C) Imaging cytometry was used to quantify GSK3β expression in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions of GSK3β mean fluorescence intensity. (D) Imaging cytometry was used to quantify GSK3β nuclear localization in primary AML (n = 86) and normal bone marrow CD34+ cells (n = 12) expressed in box-and-whisker plot displaying the median and quartile distributions. (E) Representative images of cells from the cell types indicated from imaging cytometry showing GSK3β expression and the nucleus (4′,6-diamidino-2-phenylindole [DAPI] staining). The GSK3β expression level and nuclear localization of each sample is shown. **P < .01; ***P < .001.
Interestingly, although GSK3β protein is upregulated in AML, extensive genomic studies by others reveal negligible changes in GSK3β RNA when comparing AML and normal hematopoietic cells. For example, a large microarray study of 542 AML and 72 normal hematopoietic samples reveals GSK3β RNA is merely 1.048-fold upregulated compared with normal PBMCs (supplemental Figure 2A; P < .05). This result is further supported by qPCR comparing GSK3β messenger RNA in 3 AML cell lines, 3 AML patient samples, and 3 normal patient PBMC samples, where 1-way analysis of variance reveals no significant difference between the groups (supplemental Figure 2B; P = .61992). In addition to the lack of RNA changes, no DNA mutations of GSK3β have been observed in curated AML data sets (http://cancergenome.nih.gov).
GSK3β is partially regulated through its nuclear or cytoplasmic subcellular localization.18,20 Nuclear GSK3β has access to different protein targets than the cytoplasmic pool of GSK3β leading to distinct effects on cellular processes.11,16,21 To quantify the nuclear localization of GSK3β in AML and normal controls, imaging cytometry was used. Imaging cytometry combines the high-throughput power of flow cytometry with the imaging capacity of fluorescent microscopy.22-25 By correlating the intensity of the nuclear signal to the GSK3β signal, a log2-based nuclear localization score is derived where a negative number indicates a more cytoplasmic localization and a positive number a more nuclear localization. This nuclear localization score as measured by imaging cytometry exhibits high intrasample reproducibility (supplemental Figure 3).
Imaging cytometry analysis reveals that normal CD34+ cells exhibit a predominantly cytoplasmic GSK3β localization and AML cells universally exhibit elevated nuclear localization of GSK3β (Figure 1C-E). Similar results confirming the aberrant nuclear localization of GSK3β in AML were obtained using confocal microscopy (supplemental Figure 4). Imaging cytometry was performed using primary AML samples (n = 86), AML cell lines (n = 3) and CD34+ normal hematopoietic progenitor cells (n = 12). GSK3β localization in normal CD34+ cells generated an average nuclear localization score of −0.59 ± 0.2. In contrast, AML cell lines and patient samples exhibit a dramatically increased GSK3β nuclear localization, generating an average nuclear localization score of 1.7 ± 0.045. (P < .00001) (Figure 1C). Although there is a wide distribution of GSK3β nuclear localization scores in AML, all samples tested exhibit higher scores than any of the normal CD34+ progenitor cells examined (Figure 1C).
Transformation of mouse progenitor cells by MLL-AF9 promotes GSK3β expression and nuclear localization
We next tested if leukemic transformation from normal hematopoietic progenitor cells leads to the upregulation of nuclear GSK3β using a well-characterized MLL-AF9 model that leads to transformation after ∼2 weeks of serial replating.5,17,26-30 Consistent with previous reports, normal hematopoietic cells transduced with MLL-AF9 demonstrated a marked increase in colony formation, whereas cells transduced with empty vector failed to form colonies after 2 serial replatings (supplemental Figure 5).
Similar to previous results with normal human hematopoietic cells, GSK3β expression was extremely low in the nontransformed mouse cells (Figure 2). GSK3β expression and nuclear localization increased markedly in MLL-AF9 transduced cells by the second passage. Further increases in GSK3β nuclear localization were observed after the third passage suggesting that the GSK3β nuclear localization becomes more pronounced as leukemogenesis progresses. These observations suggest GSK3β upregulation and nuclear localization can be promoted by MLL-AF9-mediated transformation and may represent an important step in AML leukemogenesis.
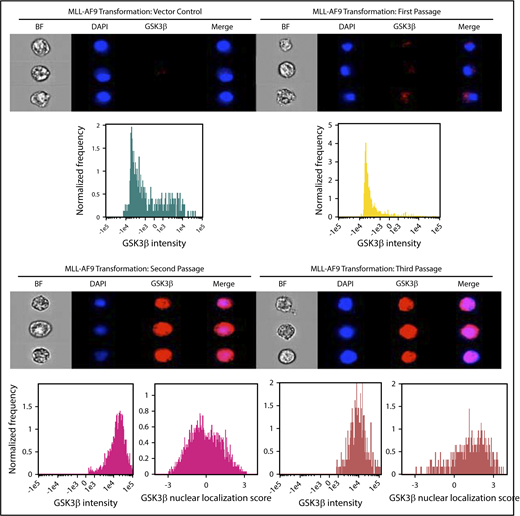
GSK3β is upregulated and exhibits high nuclear localization in mouse hematopoietic progenitor cells after MLL-AF9-mediated transformation. 5-Fluoruracil-mobilized mouse progenitor cells were transduced with vector control or MLL-AF9 and serially passaged in semisolid media for 3 passages or until colonies failed to form. Cells were analyzed by imaging cytometry following each passage for GSK3β intensity and nuclear localization score. Note: no colonies formed for vector control at the second serial passage.
GSK3β is upregulated and exhibits high nuclear localization in mouse hematopoietic progenitor cells after MLL-AF9-mediated transformation. 5-Fluoruracil-mobilized mouse progenitor cells were transduced with vector control or MLL-AF9 and serially passaged in semisolid media for 3 passages or until colonies failed to form. Cells were analyzed by imaging cytometry following each passage for GSK3β intensity and nuclear localization score. Note: no colonies formed for vector control at the second serial passage.
Nuclear GSK3β more potently promotes AML cell growth in vitro than cytoplasmic GSK3β
Nuclear GSK3β is characteristic of a wide range of AML cells, and inhibition of GSK3 impairs AML growth. This result is consistent with a growing body of literature that shows elevated nuclear GSK3β can be observed in several cancers, and subsequent inhibition or knockdown of total GSK3β inhibits cancer growth.9-15 Whether the upregulation of nuclear GSK3β specifically could promote more aggressive cancer is less clear.
To test if upregulation of nuclear GSK3β specifically promotes more aggressive AML, we knocked down endogenous GSK3β and then rescued GSK3β expression with nuclear or cytoplasmic-targeted variants in the OCI-AML3 and HL60 cell lines (supplemental Figure 6).18 Quantitation of GSK3β before and after induction suggests the amount of GSK3β expressed through the rescue system restores levels to approximately that of the parental AML cells (supplemental Figure 6). Thus, this model provides a unique tool to test if increased proportional nuclear GSK3β partitioning, not merely increased GSK3β expression, could promote more aggressive AML.
Using this rescue model we tested if nuclear or cytoplasmic GSK3β expression could promote AML colony formation. Nuclear-targeted GSK3β (NLS cells) in HL60 and OCI-AML3 cells increased colony formation by 38% ± 7.7% and 19% ± 6.1%, respectively, as compared with nontetracycline induced controls (P < .001 for HL60-NLS and P < .01 for OCI-AML3-NLS) (Figure 3A). Cytoplasmic GSK3β (Cyto cells) failed to increase colony formation (P > .05 for HL60-Cyto and OCI-AML3-Cyto) (Figure 3A). These results suggest nuclear GSK3β promotes AML growth in vitro, gaining oncogenic function compared with cytoplasmic GSK3β.
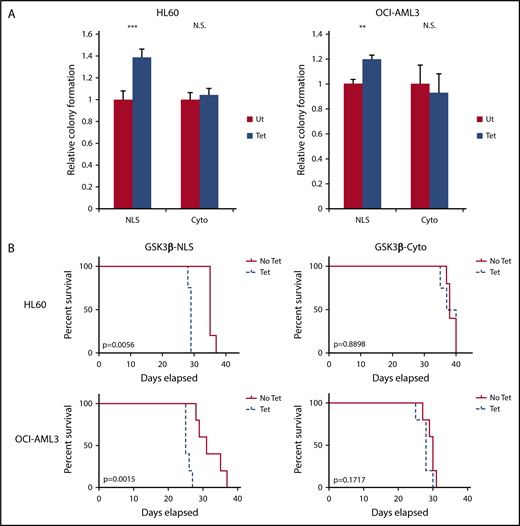
Nuclear, but not cytoplasmic, GSK3β drives AML growth. (A) The indicated cells with or without targeted GSK3β induction were cultured in semisolid media for 7 days, and relative colony formation was assessed (n = 3 or more for all groups). (B) Tetracycline-induced (0.2 μg/mL) and noninduced controls of the indicated cells were injected IV into NSG mice (n = 5 for all groups except HL60-NLS Tet, n = 4). Mice receiving tetracycline-induced cells received 100 µg/mL tetracycline supplemented water to maintain GSK3β induction in vivo. Mice were monitored for survival. **P < .01; ***P < .001. N.S., not significant.
Nuclear, but not cytoplasmic, GSK3β drives AML growth. (A) The indicated cells with or without targeted GSK3β induction were cultured in semisolid media for 7 days, and relative colony formation was assessed (n = 3 or more for all groups). (B) Tetracycline-induced (0.2 μg/mL) and noninduced controls of the indicated cells were injected IV into NSG mice (n = 5 for all groups except HL60-NLS Tet, n = 4). Mice receiving tetracycline-induced cells received 100 µg/mL tetracycline supplemented water to maintain GSK3β induction in vivo. Mice were monitored for survival. **P < .01; ***P < .001. N.S., not significant.
As nuclear GSK3β promotes AML growth in vitro, we next tested its impact in vivo. Immunodeficient mice injected with HL60 or OCI-AML3 cells were provided with tetracycline-supplemented or plain water and monitored for survival. GSK3β induction in AML cells in the mice after tetracycline-supplemented water administration was confirmed by flow cytometry (supplemental Figure 7). Nuclear, but not cytoplasmic, GSK3β led to a marked reduction in survival (survival in days: HL60-NLS, No Tet = 35, Tet = 28.75, P < .01; OCI-AML3-NLS: No Tet = 32, Tet = 25.75, P < .01) (Figure 3B). Taken together, these data suggest nuclear, but not cytoplasmic, GSK3β provides a major AML growth advantage in vivo.
Nuclear GSK3β promotes NF-κB activation
As nuclear GSK3β promotes AML growth in vivo and in vitro, we hypothesized nuclear GSK3β may impact prosurvival signaling in AML. As NF-κB is activated by GSK3β and it is a critical prosurvival factor in many cancers, we explored the connection between these factors in AML.7,11,16,31-36 Of note, nuclear GSK3β has been correlated with increased NF-κB activation in pancreatic cancer and chronic lymphocytic leukemia.11,16 Initially using a NF-κB reporter assay in 293T cells, we observed more potent NF-κB activation with nuclear GSK3β overexpression as compared with cytoplasmic (GSK3β-NLS = 18-fold induction; GSK3β-Cyto = threefold induction; P < .001) (Figure 4A; supplemental Figure 6E). Importantly, GSK3β overexpression alone can drive NF-κB dependent transcription without the necessity for additional stimuli, suggesting nuclear GSK3β may promote the basal activation of NF-κB.
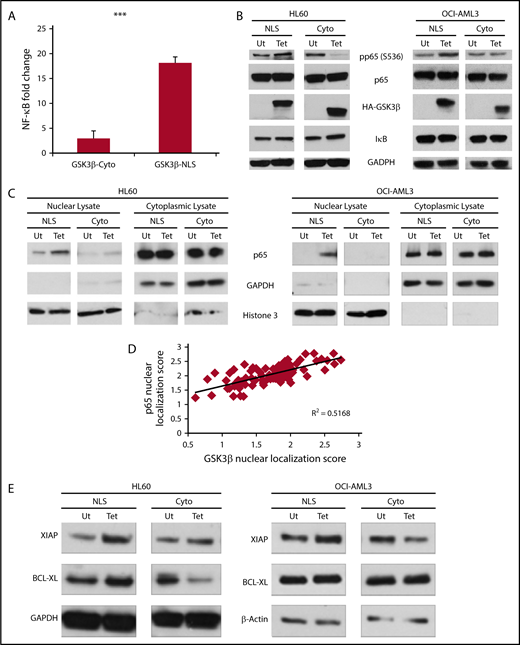
Nuclear GSK3β more potently drives NF-κB activation than cytoplasmic GSK3β. (A) 293T cells were cotransfected with an NF-κB dependent Firefly luciferase construct, a Renilla luciferase vector, and either a vector control or the indicated GSK3β construct. Forty-eight hours posttransfection, cells were analyzed for luciferase signal. Data reported are fold change from the vector control signal and is normalized for Renilla signal (n = 3). (B) The indicated cells with or without targeted GSK3β induction were analyzed for NF-κB signaling components by western blot. Note the NLS isoform is at higher molecular weight because of the construct’s exogenous NLS. (C) HL60 and OCI-AML3 cells were fractionated into nuclear and cytoplasmic lysates and analyzed for p65 nuclear translocation by western blot. (D) Linear correlation analysis of nuclear localization scores of GSK3β and p65 in primary AML samples quantified using imaging cytometry (n = 82). (E) The indicated cells with or without targeted GSK3β induction were analyzed by western blotting for changes in the NF-κB-dependent survival genes, XIAP and Bcl-XL. ***P < .001.
Nuclear GSK3β more potently drives NF-κB activation than cytoplasmic GSK3β. (A) 293T cells were cotransfected with an NF-κB dependent Firefly luciferase construct, a Renilla luciferase vector, and either a vector control or the indicated GSK3β construct. Forty-eight hours posttransfection, cells were analyzed for luciferase signal. Data reported are fold change from the vector control signal and is normalized for Renilla signal (n = 3). (B) The indicated cells with or without targeted GSK3β induction were analyzed for NF-κB signaling components by western blot. Note the NLS isoform is at higher molecular weight because of the construct’s exogenous NLS. (C) HL60 and OCI-AML3 cells were fractionated into nuclear and cytoplasmic lysates and analyzed for p65 nuclear translocation by western blot. (D) Linear correlation analysis of nuclear localization scores of GSK3β and p65 in primary AML samples quantified using imaging cytometry (n = 82). (E) The indicated cells with or without targeted GSK3β induction were analyzed by western blotting for changes in the NF-κB-dependent survival genes, XIAP and Bcl-XL. ***P < .001.
We next tested if nuclear GSK3β could better activate NF-κB in AML than cytoplasmic GSK3β. The induction of nuclear, but not cytoplasmic, GSK3β led to increased activation of the p65 subunit as evidenced by increased phosphorylation at serine 536 (S536) and nuclear translocation of p65 (Figure 4B-C). GSK3β expression did not promote IκB degradation, suggesting GSK3β acts distal to the IκB complex in AML (Figure 4B).11,16
If nuclear GSK3β enhances the nuclear localization of p65 in AML, then GSK3β and p65 nuclear localization should positively correlate in AML patient samples. Linear regression analysis of GSK3β and p65 nuclear localization scores from our ECOG-ACRIN cohort revealed a positive correlation (R2 = 0.5168, P < .001) suggesting the localization of these proteins is related in AML (Figure 4D). This result suggests that nuclear GSK3β may influence the accumulation of p65 in the nucleus where it can promote transcription of NF-κB target genes.
Finally, given that nuclear GSK3β activates NF-κB, we next assessed for changes in NF-κB-dependent target genes that are involved in AML survival and drug resistance. Nuclear, but not cytoplasmic, GSK3β increased the expression of the prosurvival gene Bcl-XL in HL60 cells as well as the expression of XIAP in both HL60 and OCI-AML3 cells (Figure 4E). In contrast, cytoplasmic GSK3β expression led to a reduction of Bcl-XL (HL60) and XIAP (OCI-AML3) (Figure 4E).
Nuclear GSK3β protects against daunorubicin-mediated AML cell killing in vitro and in vivo
Because nuclear GSK3β promotes prosurvival NF-κB signaling, which is associated with AML drug resistance,35,36 we tested if nuclear GSK3β promotes AML drug resistance. GSK3β-NLS protected AML cells from daunorubicin-mediated cell killing doubling the median lethal dose (LD50) from 197 nM to 400 nM while GSK3β-Cyto lowered the daunorubicin LD50 from 247 nM to 185 nM (Figure 5A). The protective effect of nuclear GSK3β on AML cell survival was found to be expression dependent as increasing amounts of nuclear GSK3β increased drug resistance (Figure 5B). Similar results were observed utilizing another AML therapeutic, cytarabine (Figure 5B).
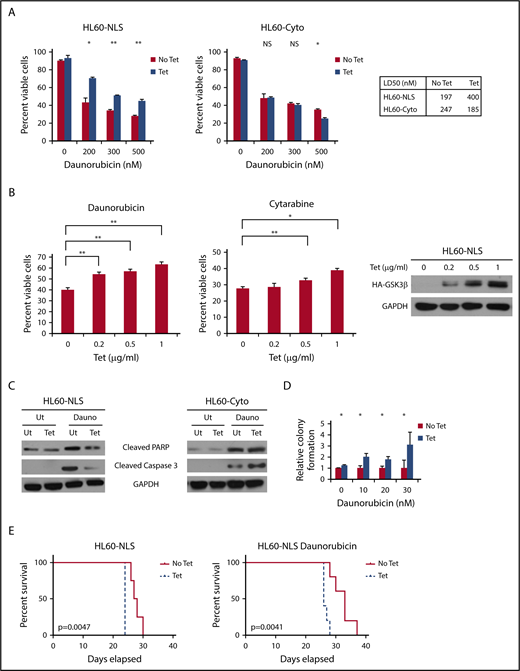
Nuclear localization of GSK3β promotes resistance to standard AML chemotherapeutics. (A) The indicated cells with or without targeted GSK3β induction were treated with various doses of daunorubicin for 24 hours, and cell viability was determined by trypan blue staining (n = 2, HL60-NLS; n = 4, HL60-Cyto). (B) HL60-NLS cells were treated with tetracycline and 200 nM daunorubicin or 25 nM cytarabine for 2 days. The cell viability was assessed by Hoescht staining (n = 3; *P < .05, **P < .01). (C) The indicated cells with or without targeted GSK3β induction were treated with daunorubicin (200 nM) for 18 hours and analyzed by western blot for PARP or caspase 3 cleavage. (D) The indicated cells with or without targeted GSK3β induction were treated with daunorubicin for 2 days. Daunorubicin was washed away, the cells were plated in semisolid media, and colony formation was assessed after 7 days (n = 2). (E) Tetracycline-induced (0.2 µg/mL) and noninduced HL60-NLS cells were injected IV into NSG mice. Mice receiving tetracycline-induced cells were provided tetracycline-supplemented water (100 µg/mL). Four days post–cell injection, mice were injected with 1.5 mg/kg daunorubicin once a day for 3 days and monitored for survival (n = 5).
Nuclear localization of GSK3β promotes resistance to standard AML chemotherapeutics. (A) The indicated cells with or without targeted GSK3β induction were treated with various doses of daunorubicin for 24 hours, and cell viability was determined by trypan blue staining (n = 2, HL60-NLS; n = 4, HL60-Cyto). (B) HL60-NLS cells were treated with tetracycline and 200 nM daunorubicin or 25 nM cytarabine for 2 days. The cell viability was assessed by Hoescht staining (n = 3; *P < .05, **P < .01). (C) The indicated cells with or without targeted GSK3β induction were treated with daunorubicin (200 nM) for 18 hours and analyzed by western blot for PARP or caspase 3 cleavage. (D) The indicated cells with or without targeted GSK3β induction were treated with daunorubicin for 2 days. Daunorubicin was washed away, the cells were plated in semisolid media, and colony formation was assessed after 7 days (n = 2). (E) Tetracycline-induced (0.2 µg/mL) and noninduced HL60-NLS cells were injected IV into NSG mice. Mice receiving tetracycline-induced cells were provided tetracycline-supplemented water (100 µg/mL). Four days post–cell injection, mice were injected with 1.5 mg/kg daunorubicin once a day for 3 days and monitored for survival (n = 5).
To further characterize drug resistance effects of nuclear GSK3β, we tested if nuclear GSK3β inhibited apoptosis. Western blot analysis of AML cells expressing GSK3β-NLS revealed significant decreases in the cleavage of the apoptotic mediators caspase 3 and PARP after daunorubicin treatment as compared with control cells (Figure 5C). Additionally, the expression of GSK3β-Cyto promotes cleavage of caspase 3 in response to daunorubicin, suggesting cytoplasmic GSK3β may promote apoptosis instead of impairing it.
In addition to promoting resistance to daunorubicin-mediated killing, nuclear GSK3β protects colony formation in HL60 cells treated with low doses of daunorubicin. For example, nuclear GSK3β induction led to threefold more colonies as compared with control cells after treatment with 30 nM daunorubicin (P < .05) (Figure 5D).
GSK3β-NLS also promotes AML daunorubicin resistance in mice. For this model, immunodeficient mice were injected with HL60-NLS cells, and the mice were provided either tetracycline-supplemented water to induce nuclear GSK3β or plain water. Mice were treated with vehicle or daunorubicin and assessed for survival. As expected, the GSK3β-NLS promoted more aggressive disease regardless of the presence of daunorubicin. Interestingly, although treatment with daunorubicin improved survival, the induction of nuclear GSK3β significantly limited daunorubicin’s prosurvival effects, suggesting nuclear GSK3β promotes daunorubicin resistance in vivo (average days survival: No Tet/dauno = 32, Tet/dauno = 26.6; No Tet = 27.75, Tet = 24; P < .01 for each No Tet/Tet comparison) (Figure 5E).
GSK3β and p65 nuclear localization correlate to poorer patient survival
As GSK3β-NLS promotes drug resistance and most AML patients die of drug resistant disease, we hypothesized that GSK3β nuclear localization will predict patient survival.37,38 Although the nuclear localization of GSK3β is universally elevated in AML, there is a wide range in patients, suggesting it may serve as a useful biomarker (Figure 1C-D). We also assessed the nuclear localization of the related protein, p65. Similar to GSK3β, p65 also exhibits a wide range of nuclear localization scores among AML patients (Figure 6A).
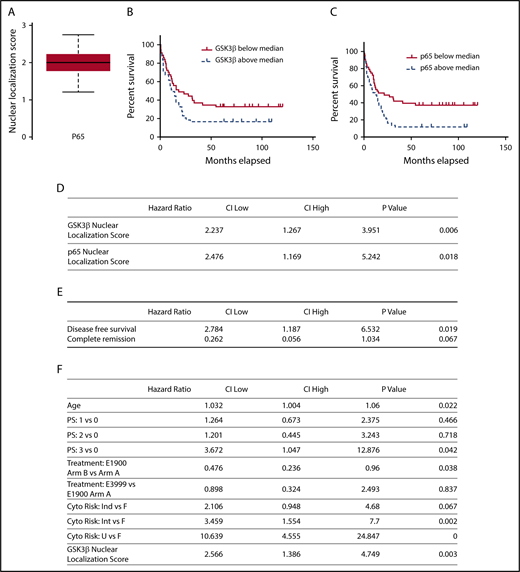
Nuclear localization of GSK3β and p65 predict AML survival. (A) Nuclear localization scores of p65 were quantified using imaging cytometry (n = 86). A box-and-whisker plot displaying the median and quartile distributions is shown. (B-C) Kaplan-Meier survival curve analysis comparing overall survival of AML patients based on nuclear localization scores of GSK3β (B) or p65 (C) above and below the median. (D) Nuclear localization scores of GSK3β and p65 were correlated to patient survival using multivariate Cox proportional hazard analysis controlling for age, sex, treatment group, and performance status (n = 86). Nuclear GSK3β localization improves a multivariate Cox proportional hazards model for overall survival. (E) Nuclear localization scores of GSK3β were correlated to patient complete remission and disease-free survival using a logistic regression model (n = 86). Increased GSK3β is associated with less DFS and trended to less CR (HR = 2.784 and 0.262, P = .019 and 0.067, respectively). (F) Analysis controlling for age, performance status, and treatment was performed both without and with GSK3β (top and bottom panels, respectively). The likelihood ratio is 9.1 (degrees of freedom = 1, P = .0026, n = 86) indicating the model including GSK3β as a covariate significantly improves the model fit to the data.
Nuclear localization of GSK3β and p65 predict AML survival. (A) Nuclear localization scores of p65 were quantified using imaging cytometry (n = 86). A box-and-whisker plot displaying the median and quartile distributions is shown. (B-C) Kaplan-Meier survival curve analysis comparing overall survival of AML patients based on nuclear localization scores of GSK3β (B) or p65 (C) above and below the median. (D) Nuclear localization scores of GSK3β and p65 were correlated to patient survival using multivariate Cox proportional hazard analysis controlling for age, sex, treatment group, and performance status (n = 86). Nuclear GSK3β localization improves a multivariate Cox proportional hazards model for overall survival. (E) Nuclear localization scores of GSK3β were correlated to patient complete remission and disease-free survival using a logistic regression model (n = 86). Increased GSK3β is associated with less DFS and trended to less CR (HR = 2.784 and 0.262, P = .019 and 0.067, respectively). (F) Analysis controlling for age, performance status, and treatment was performed both without and with GSK3β (top and bottom panels, respectively). The likelihood ratio is 9.1 (degrees of freedom = 1, P = .0026, n = 86) indicating the model including GSK3β as a covariate significantly improves the model fit to the data.
We next used a Cox proportional hazard model to test if increased p65 and GSK3β nuclear localization scores correlate to poorer patient survival in the ECOG-ACRIN cohort, revealing that increased GSK3β or p65 nuclear localization strongly correlates with decreased patient survival (GSK3β hazard ratio [HR] = 2.237, P < .01; p65 HR = 2.476, P < .05) (Figure 6D). We also individually correlated total expression of GSK3β, p65, β-catenin, and pS9 GSK3β to patient survival. Interestingly, none of these parameters correlated to poorer patient survival suggesting the importance of the nuclear pool of these proteins.
Given that GSK3β nuclear localization correlates with poorer patient outcomes, we characterized GSK3β nuclear localization within the major AML cytogenetic risk groups. These risk groups provide important prognostic information and are widely used to guide treatment decisions.2,39,40 There was no statistically significant difference in GSK3β nuclear localization between patients in these groups, suggesting that GSK3β nuclear localization is not merely characteristic of 1 cytogenetic risk group (supplemental Table 2).
Given that increased nuclear GSK3β is associated with poorer overall survival and our in vivo and in vitro models suggest nuclear GSK3β may promote drug resistance, we used a logistic regression model to test if increased nuclear GSK3β is associated with lower rates of complete remission (CR) and decreased disease-free survival (DFS). Increased nuclear GSK3β was associated with less DFS (Figure 6E; HR = 2.784, P = .019). Increased nuclear GSK3β also trended toward a statistically significant association with decreased CR (Figure 6E; HR = 0.262, P = .067). Taken together, these data further support the association of nuclear GSK3β with worse clinical outcomes.
We next developed a multivariate Cox proportional hazard model that included cytogenetic risk group, age, performance status, and treatment received to test if nuclear GSK3β localization score could improve current prognostic models. Inclusion of GSK3β nuclear localization yielded a high HR (HR = 2.566, P = .003) and significantly improved the fit of this multivariate model, suggesting nuclear GSK3β may function as a useful AML biomarker (likelihood ratio = 9.1, P < .01) (Figure 6F).
To further test if GSK3β and p65 nuclear localization score can be used as AML biomarkers, we compared overall survival between patients with low and high GSK3β or p65 nuclear localization scores using the median scores as the cutoff. Patients with GSK3β or p65 nuclear localization scores above the median exhibited worse overall survival than patients below the median (GSK3β HR = 1.637, P = .008; p65 HR = 1.948, P = .048) (Figure 6B-C). The major impact of nuclear GSK3β localization score on AML survival, taken together with the cell and animal based studies, suggests elevated nuclear GSK3β localization can directly promote poorer patient survival. As the primary determinant of AML patient survival is drug resistance/refractoriness, these biomarker studies are also suggestive that nuclear GSK3β promotes AML drug resistance in patients.
Discussion
In general, cancers are driven by genetic and epigenetic changes that promote cell survival. For example, mutation of the AML oncogene, NPM1, leads to aberrant cytoplasmic localization and altered function.41 Here we report a novel oncogenic mechanism where aberrant nuclear localization of wild-type GSK3β can drive AML. To our knowledge, no GSK3β mutations or significant transcriptional changes have been reported in AML, and our present qPCR data further enforce that conclusion (supplemental Figure 2; http://cancergenome.nih.gov).1
Our studies demonstrate that AML exhibits a high level of expression and aberrant nuclear localization of GSK3β in a range of genetic subtypes. As AML is highly heterogeneous, this finding strongly suggests that the aberrant localization of GSK3β may represent a more general AML hallmark and therefore common therapeutic target in AML.
Our model provides a unique tool to dynamically show increased nuclear GSK3β promotes more aggressive, drug resistant AML, producing similar results in 2 different AML cell lines. Drug resistance in AML is a major clinical challenge because the majority of patients relapse with drug-resistant disease.39 Although we recognize the absolute quantification of GSK3β in our model is difficult to correlate directly to a clinical context, we argue our model provides foundational evidence that nuclear GSK3β more potently promotes AML growth and drug resistance than cytoplasmic GSK3β. This increased relative potency is appreciated in our clinical data. Indeed, independent of overall GSK3β expression levels, a higher proportion of nuclear to cytoplasmic GSK3β, reflected in the nuclear localization coefficient, correlated to poorer patient survival in 86 clinically annotated samples collected from multiple centers. Interestingly, although patients with elevated nuclear GSK3β exhibited poorer overall survival and DFS, the correlation with CR only trended to significance. This suggests that increased nuclear GSK3β may correlate more with drug resistance that drives relapse rather than an initial treatment failure. Previous work suggests the AML stem cell population as a potential cause of relapse and given the key role of GSK3β in MLL leukemogenesis and the importance of GSK3β’s main target β-catenin in AML stem cells, further studies with nuclear GSK3β and AML stem cells are of interest.
Interestingly in addition to GSK3β nuclear localization, p65 nuclear localization also correlates to poor patient survival. This finding follows from the observation that GSK3β and p65 nuclear localization correlate. As p65 nuclear translocation represents a key step in NF-κB activation, patients with poorer survival may exhibit more active NF-κB signaling. Although it is well-established that NF-κB promotes cancer drug resistance, to our knowledge this is the first report of p65 nuclear localization correlating to AML patient survival.
Although GSK3β protein expression is markedly upregulated in AML, it did not correlate to poorer patient survival. This suggests the relative nuclear localization of GSK3β is the key factor and fits with our observations that nuclear, but not cytoplasmic, GSK3β leads to AML resistance to chemotherapy. This observation is consistent with previous studies that have suggested that cytoplasmic GSK3β can exhibit proapoptotic effects in other disease models.20,42-44 Thus, the overall effect of GSK3β in AML may be dependent on the relative balance between proapoptotic cytoplasmic GSK3β and oncogenic nuclear GSK3β.
Targeting GSK3 is a highly desirable therapeutic approach that inhibits AML cell growth while promoting normal hematopoietic stem cell growth.3,8,45 As normal cells lack an appreciable pool of nuclear GSK3β, our study sheds light on why GSK3 inhibitors have such different biological effects on normal and AML cells. Furthermore, our data suggest that specifically targeting the nuclear pool of GSK3β may be of specific therapeutic interest.
GSK3β is known to activate the transcription factor NF-κB, a key factor of cancer cell survival and drug resistance.7,31,35,36,46 In AML, nuclear GSK3β more robustly activated NF-κB through p65 phosphorylation and nuclear translocation leading to increased expression of the NF-κB prosurvival genes Bcl-XL and XIAP. Interestingly, we observed a strong correlation of p65 and GSK3β nuclear localization in a large set of patient samples, further supporting a relationship between these factors. As GSK3β is a central kinase that is known to regulate numerous nuclear transcription factors such as CREB, Fos/Jun, NFAT, p53, and C/EBP, nuclear GSK3β likely exhibits additional oncogenic effects beyond NF-κB in AML.6,42
At first glance, our observation that nuclear GSK3β directly promotes AML is paradoxical given that β-catenin has been shown to be crucial for AML initiation in mouse models and GSK3β is known to impair the function and lead to the degradation of β-catenin. Although β-catenin is required for AML formation, it is not required for established AML.22 Furthermore, we and others have shown GSK3β affects AML through β-catenin-independent mechanisms.3-5 Although careful regulation of GSK3β is necessary to maintain adequate β-catenin signaling, our data suggest that in established AML there is overall a selection for cells exhibiting elevated nuclear GSK3β and this elevated nuclear GSK3β promotes more aggressive AML. Future studies will be necessary to determine the role of nuclear and cytoplasmic GSK3β on β-catenin signaling during AML leukemogenesis.
Interestingly, previous studies in AML have yielded contradictory results about the role of GSK3β in AML that may be explained by its differential roles in leukemogenesis and disease progression. One recent study suggests GSK3β deletion in normal hematopoietic stem cells promotes a myleodysplastic syndromelike state, which eventually progresses to aggressive AML.47 It is possible that early accentuation of β-catenin signaling may promote the initial AML transformation. However, many groups have shown that genetic and pharmacologic inhibition of GSK3β impair the growth of established AML.3-5 Our study helps reconcile these previous studies by defining a specific role of nuclear GSK3β in driving AML after initial transformation.
Our work suggests that 1 novel strategy to specifically target AML cells may involve directly targeting the nuclear pool of GSK3β. The nuclear translocation of GSK3β has been reported to involve the combination of regulatory phosphorylation at the S9 residue as well as interaction with scaffolding proteins such as Axin and Frat1, promoting and opposing GSK3β nuclear localization, respectively.18,21,48,49 Future therapeutic strategies could involve targeting molecules involved in GSK3β transport to promote nuclear export. In addition to developing a potential therapeutic strategy, studies of GSK3β nuclear transport in AML are important as it is likely that 1 or more components of this pathway are dysregulated in AML leading to high levels of nuclear localization observed.
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported by the Cytometry and Imaging Microscopy Core, Athymic Animal Core, and Hematopoietic Biorepository Core of the Case Comprehensive Cancer Center (National Cancer Institute, National Institutes of Health grant P30CA043703). This work was also supported by the Leukemia and Lymphoma Society, American Cancer Society (D.N.W.), and a National Cancer Institute, National Institutes of Health training award (F30CA18466) (J.J.I.-H.). The biomarker study was coordinated by the ECOG-ACRIN Cancer Research Group (Robert L. Comis and Mitchell D. Schnall, Group Cochairs) and supported by the National Cancer Institute, National Institutes of Health under the following award numbers: CA180820, CA21115, CA180794, CA23318, CA66636, CA180853, CA14548, CA180828, CA73590, CA180795, CA49883, CA11083, CA189859, CA14958, CA180791, and CA18079.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Authorship
Contribution: J.J.I.-H. and D.N.W. designed the research; J.J.I.-H., I.K.G., N.M.M., A.J.R., and M.U. performed experiments; J.J.I.-H., I.K.G., M.d.L., H.M.L., V.W., E.P., A.J.R., and D.N.W. analyzed and interpreted data; J.J.I.-H. and D.N.W. wrote the manuscript; H.F., L.C., and M.T. ran the ECOG clinical trial from which the patient samples were obtained; and D.N.W. funded the research.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: David N. Wald, Case Western Reserve University, 2103 Cornell Rd, Cleveland, OH 44106; e-mail: dnw@case.edu.

