

Key Points
biCEBPA-mutated AML is a heterogeneous subentity that consists of at least 2 subgroups with different genetic and clinical features.

Abstract
Biallelic mutations of the CCAAT/enhancer binding protein α (CEBPA) gene define a distinct genetic entity of acute myeloid leukemia (AML) with favorable prognosis. The presence of GATA2 and CSF3R mutations that are specifically associated with this subgroup but not mutated in all samples suggests a genetic heterogeneity of biCEBPA-mutated AML. We characterized the mutational landscape of CEBPA-mutated cytogenetically normal AML by targeted amplicon resequencing. We analyzed 48 biallelically mutated CEBPA (biCEBPA), 32 monoallelically mutated CEBPA (moCEBPA), and 287 wild-type CEBPA (wtCEBPA) patient samples from German AML Cooperative Group studies or registry. Targeted sequencing of 42 genes revealed that moCEBPA patients had significantly more additional mutations and additional mutated genes than biCEBPA patients. Within the group of biCEBPA patients, we identified 2 genetic subgroups defined by the presence or absence of mutations in chromatin/DNA modifiers (C), cohesin complex (C), and splicing (S) genes: biCEBPACCSpos (25/48 [52%]) and biCEBPACCSneg (23/48 [48%]). Equivalent subgroups were identified in 51 biCEBPA patients from the Cancer Genome Project. Patients in the biCEBPACCSpos group were significantly older and had poorer overall survival and lower complete remission rates following intensive chemotherapy regimens compared with patients in the biCEBPACCSneg group. Patients with available remission samples from the biCEBPACCSpos group cleared the biCEBPA mutations, but most had persisting CCS mutations in complete remission, suggesting the presence of a preleukemic clone. In conclusion, CCS mutations define a distinct biological subgroup of biCEBPA AML that might refine prognostic classification of AML. This trial was registered at www.clinicaltrials.gov as #NCT00266136 and NCT01382147.
Introduction
Mutations in the CCAAT/enhancer binding protein α (CEBPA) gene are detected in ∼10% of patients with cytogenetically normal (CN) acute myeloid leukemia (AML). CEBPA mutations can be either biallelic or monoallelic. Most patients with 2 CEBPA mutations carry 1 frameshift mutation in the N-terminal part of the protein and the other one in the bZIP domain, which is located at the C terminus.1,2 A single CEBPA mutation is most often found at the N terminus. N-terminal frameshift mutations specifically abolish the translation of the full-length (42-kDa) protein of CEBPA, leading to the overexpression of a shorter, dominant-negative 30-kDa isoform of CEBPA.3 C-terminal in-frame mutations disrupt the homo- and heterodimerization domains and therefore impair the DNA-binding activity of the CEBPA protein.4,5 Only patients with biallelically mutated CEBPA (biCEBPA) have favorable outcomes when compared with other CN-AML patients.2,6-12 Because of its unique characteristics, AML with biCEBPA mutations are classified as a distinct entity in the 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia.10 biCEBPA mutations are rarely associated with other prognostic mutations such like internal tandem duplications (ITD) of the fms-like tyrosine kinase 3 (FLT3) gene, mutations in the tyrosine kinase domain (TKD) of FLT3, mutations in nucleophosmin 1 gene (NPM1), or partial tandem duplications of the lysine methyltransferase 2A (KMT2A) gene. We previously identified a specific association of biCEBPA mutations with mutations in the transcription factor GATA2 in 39% of cases.13 CSF3R mutations are described to be also frequently mutated (29%) in biCEBPA patients.14 In the present study, we aimed to characterize the mutational spectrum of moCEBPA and biCEBPA CN-AML patients. We also analyzed outcome in patients with biCEBPA mutations based on these additional mutations.
Methods
Targeted amplicon resequencing (Agilent Haloplex; target region, ∼62 kbp) was used to analyze 42 genes and hotspots in 80 CEBPA mutated AML patients (32 monoallelically mutated CEBPA [moCEBPA] and 48 biCEBPA); 65 of these AML patients (40 biCEBPA and 25 moCEBPA) were enrolled in the German AML Cooperative Group (AMLCG) 1999 or 2008 multicenter randomized phase 3 trials or in the AMLCG registry. All other patients were treated according to standard intensive induction/postremission protocols. Long-term follow-up data were available for all moCEBPA patients and 45 of 48 biCEBPA patients.
Treatment protocols of the AMLCG 1999 (registered at www.clinicaltrials.gov as #NCT00266136) or AMLCG 2008 (#NCT01382147) trials have been reported previously.15,16 Study protocols were approved by the ethics committees of the participating centers. From all patients, written informed consent to the scientific use of surplus samples was obtained in accordance with the Declaration of Helsinki.
Genomic DNA was extracted from bone marrow or peripheral blood samples using QIAcube technology (QIAGEN, Hilden, Germany). 200 ng double-stranded genomic DNA, as quantified by a Qubit Fluorometer 2.0 (Life Technologies, Carlsbad, CA), was used for the target capture reaction from each sample. A custom-design HaloPlex Target Enrichment kit (1-500 kb; Agilent, Boeblingen, Germany) was employed to capture the target regions according to the HaloPlex Target Enrichment System-Fast Protocol Version B. Paired-end sequencing (2 × 250 bp) was performed on an Illumina MiSeq instrument (Illumina, San Diego, CA). Sequence alignment and variant detection was performed as described previously.17 A variant allele frequency threshold of 2% was set for mutation detection. CEBPA mutations in diagnosis and if available in remission samples were identified using fragment-length analysis with subsequent Sanger sequencing.1 FLT3-ITD status was analyzed via fragment length analysis.18 CSF3R (NM_156039) exons 14-17 were analyzed by Sanger sequencing.19 Sequence traces were analyzed with Sequencher software (Gene Codes, Ann Arbor, MI). Target regions of remission samples were analyzed by sequencing analysis on the Ion PGM system (coverage ∼1000-fold).
Differences in patient characteristics were calculated using the Fisher’s exact test for categorical variables and Mann-Whitney U test for continuous variables. The Fisher’s exact test was used to compare pairwise mutation frequencies among the 3 CEBPA groups (moCEBPA or biCEBPA and wild-type CEBPA [wtCEBPA]) for genes with a mutation frequency ≥5% in at least 1 of the CEBPA groups. Adjustment for multiple hypothesis testing was performed using the method described by Benjamini and Hochberg.20 To evaluate differences in additional mutated genes or number of additional gene mutations between moCEBPA and biCEBPA patients, we used the Mann-Whitney U test. We further characterized the mutation profile of biCEBPA-mutated patients. There is evidence that AML patients with mutations in chromatin modifier and splicing genes have poor outcomes.21 We therefore analyzed outcomes depending on presence and absence of chromatin modifiers and splicing genes and added cohesion genes to this group upon its interaction with chromatin. All clinical end points were defined according to generally accepted criteria.22 Overall survival (OS) was calculated from the date of randomization for patients in clinical trials or at the date of first diagnosis for patients in registry until death from any cause. Relapse-free survival (RFS) was determined for responders from the first day of complete remission (CR) until relapse or death from any cause. Estimated probabilities of OS and RFS were calculated using the Kaplan-Meier method. The log-rank test evaluated statistical differences (P value) between survival distributions. Patients who had undergone allogeneic bone marrow transplantation were censored at the time of transplantation. For all tests, P ≤ .05 was considered significant. Statistical computations were performed using SPSS software version 20.0 (SPSS, Chicago, IL) and the R software package version 3.3.1 (R Foundation for Statistical Computing; www.r-project.org).
Results
Using targeted resequencing, we analyzed a cohort of 80 CEBPA-mutated CN-AML patients (n = 48 biCEBPA, n = 32 moCEBPA) for mutations in 42 genes (supplemental Tables 1 and 2). CEBPA and FLT3-ITD mutational status was analyzed using conventional techniques (supplemental Tables 3 and 4).
We evaluated the mutational spectrum of our CEBPA cohort in comparison with a cohort of 287 wtCEBPA patients.17 Among the 20 genes with a mutation frequency of ≥5%, 8 were significantly associated with CEBPA mutation status (Figure 1).
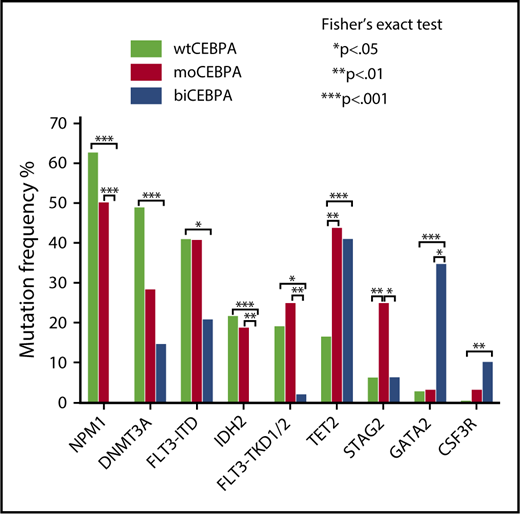
Mutation spectrum of moCEBPA, biCEBPA, and wtCEBPA. Evaluation of the mutation spectrum of moCEBPA (n = 32) and biCEBPA (n = 48) patients in comparison with wtCEBPA samples (n = 287). Eight of 20 genes with a mutation frequency of ≥5% were significantly associated with ≥1 groups.
Mutation spectrum of moCEBPA, biCEBPA, and wtCEBPA. Evaluation of the mutation spectrum of moCEBPA (n = 32) and biCEBPA (n = 48) patients in comparison with wtCEBPA samples (n = 287). Eight of 20 genes with a mutation frequency of ≥5% were significantly associated with ≥1 groups.
We found significantly more STAG2 (25%; P = .04) and FLT3-TKD1/2 (25%; P = .01) mutations in the moCEBPA subgroup than in the biCEBPA group. Most of the TKD mutations in the moCEBPA cohort were subclonal. GATA2 (35%) and CSF3R (10%) mutations were most frequently found in biCEBPA-mutated patient samples. In our cohort, IDH2 and NPM1 mutations were mutually exclusive of biCEBPA mutations.
DNMT3A was most frequently detected in wtCEBPA (49%) patients. The frequency of DNMT3A mutations in wtCEBPA was significantly different from those in biCEBPA (15%; P < .001), but not moCEBPA (28%; P = .07).
We found a higher frequency of FLT3-ITD mutations in moCEBPA (41%; P = .13) and wtCEBPA (41%; P = .02) patients than biCEBPA patients (21%).
TET2 mutations were significantly more frequent in moCEBPA (44%, P = .003) and biCEBPA (42%, P = .001) patients than wtCEBPA patients. In our data set, the frequency of TET2 mutations is much higher than that of other CEBPA-mutated AML cohorts.23 The frequency in our CEBPA cohort is also higher when compared with other AML cohorts.24,25
TET2 mutations were located within the conserved region of the gene and are thus likely pathogenic.26,27 These data show that TET2 mutations define a subgroup of CEBPA-mutated samples regardless of the CEBPA allelic mutation status.
Next, we compared both CEBPA subgroups. With the exception of sex, there were no significant differences in baseline characteristics between moCEBPA and biCEBPA patient samples (Table 1).
Characteristics of moCEBPA vs biCEBPA patients
| Characteristic | moCEBPA | biCEBPA | P |
|---|---|---|---|
| Age, y | .69 | ||
| Median | 62 | 57 | |
| Range | 16-78 | 20-84 | |
| Sex | |||
| Female | 23 (72) | 23 (48) | .04 |
| WBC count, ×109/L | 31/32 | 47/48 | .47 |
| Median | 30 | 28 | |
| Range | 0.4-188 | 1.3-408.6 | |
| Hemoglobin, g/dL | 29/32 | 44/48 | .13 |
| Median | 9.1 | 10 | |
| Range | 2.7-13.2 | 6.4-13.8 | |
| Platelet count, ×109/L | 28/32 | 47/48 | .09 |
| Median | 52 | 32 | |
| Range | 3-291 | 3-151 | |
| Bone marrow blasts, % | 19/32 | 40/48 | .65 |
| Median | 85 | 71 | |
| Range | 30-95 | 20-100 | |
| LDH, U/dL | 20/32 | 36/48 | .67 |
| Median | 392 | 479 | |
| Range | 152-2666 | 205-2510 |
| Characteristic | moCEBPA | biCEBPA | P |
|---|---|---|---|
| Age, y | .69 | ||
| Median | 62 | 57 | |
| Range | 16-78 | 20-84 | |
| Sex | |||
| Female | 23 (72) | 23 (48) | .04 |
| WBC count, ×109/L | 31/32 | 47/48 | .47 |
| Median | 30 | 28 | |
| Range | 0.4-188 | 1.3-408.6 | |
| Hemoglobin, g/dL | 29/32 | 44/48 | .13 |
| Median | 9.1 | 10 | |
| Range | 2.7-13.2 | 6.4-13.8 | |
| Platelet count, ×109/L | 28/32 | 47/48 | .09 |
| Median | 52 | 32 | |
| Range | 3-291 | 3-151 | |
| Bone marrow blasts, % | 19/32 | 40/48 | .65 |
| Median | 85 | 71 | |
| Range | 30-95 | 20-100 | |
| LDH, U/dL | 20/32 | 36/48 | .67 |
| Median | 392 | 479 | |
| Range | 152-2666 | 205-2510 |
Data represent number or n (%) of patients, unless otherwise indicated. Bold indicates significant P values.
LDH, lactate dehydrogenase; WBC, white blood cell.
Targeted sequencing analysis of these 42 genes revealed that moCEBPA patients had significantly more additional mutations than biCEBPA patients (mean: 4.0 ± 1.7 vs 2.2 ± 1.5; P < .001). The number of additional mutated genes was also significantly higher in moCEBPA patients than biCEBPA patients (mean: 3.7 ± 1.6 vs 2.0 ± 1.3; P < .001) (supplemental Figure 1). The 2 groups differed significantly with regard to mutations in the following genes: NPM1, FLT3-TKD1/2, IDH2, STAG2, and GATA2 (supplemental Figure 2). Patients without any additional mutations were more frequently found in the biCEBPA group than in the moCEBPA cohort (6/48, 13% vs 1/32, 3%; P = .23; Figure 2).
Moreover, we identified 2 distinct genetic subgroups in biCEBPA patients based on comutated genes (Figure 2). biCEBPA patients with mutations in chromatin/DNA modifiers (C), cohesin complex (C), and splicing (S) genes were defined as biCEBPACCSpos (n = 25). The biCEBPACCSpos group is mainly defined by TET2 (20/25), DNMT3A (7/25), and WT1 (7/25) mutations. The other group, biCEBPACCSneg, includes biCEBPA patients with mutations in signaling factors only and/or GATA2 and patients with no additional mutations in the 42 genes analyzed. In a cohort of 51 biCEBPA patients without recurrent chromosomal abnormalities and genomic rearrangements (analyzed in a large AML cohort of 1540 patients reported by the Cancer Genome Project), we also identified a biCEBPACCSneg subgroup; however, this group is smaller (31%) than ours (48%) (supplemental Figure 3). The most frequently mutated gene in the biCEBPACCSpos group of the Cancer Genome Project was WT1 (20%).21
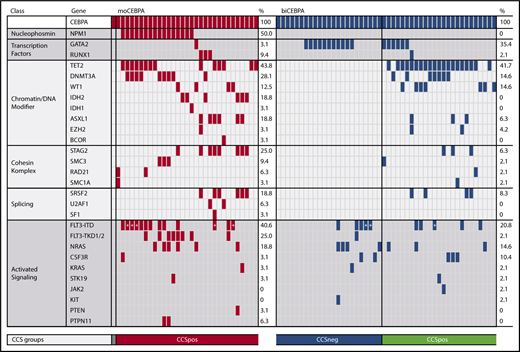
Frequency of genetic alterations organized by categories of related genes and genetic groups. The heatmap includes all genes that were mutated in either the moCEBPA (red) or biCEBPA (dark blue) subgroup. biCEBPA patient samples were further separated in 2 biological groups: CCSneg (blue) and CCSpos (green). Patients with a signal ratio of FLT3-ITD ≥0.5 are marked with an asterisk.
Frequency of genetic alterations organized by categories of related genes and genetic groups. The heatmap includes all genes that were mutated in either the moCEBPA (red) or biCEBPA (dark blue) subgroup. biCEBPA patient samples were further separated in 2 biological groups: CCSneg (blue) and CCSpos (green). Patients with a signal ratio of FLT3-ITD ≥0.5 are marked with an asterisk.
biCEBPACCSpos patients were significantly older than biCEBPACCSneg patients (66 vs 48 years, P = .005; Figure 3A; Table 2). Patients in CCSpos subgroup also had a lower CR rate (Figure 3B) and a higher early death rate (supplemental Table 6).
![Figure 3. Age and CR rate of biCEBPACCSpos, biCEBPACCSneg, and moCEBPA patients. (A) Age of biCEBPACCSneg (median = 48), biCEBPACCSpos (median = 66), and moCEBPA (median = 62) patients. Scatter dot plot, median with interquartile range. (B) The CR rate was significantly higher in biCEBPACCSneg patients (91% [20/22]) than biCEBPACCSpos (61% [14/23]; P = .035) and moCEBPA patients (P = .027).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/20/10.1182_bloodadvances.2018016840/4/m_advances016840f3.png?Expires=1769092458&Signature=Zjz7VDdnv~yoOOC4mKgMJwX4Yt6zMmwQg--o-N2Sg5ssAt7Va~AM6Fxv9zHlNyx9I7rMvPKT0Sl4KarL2Nw2xUUKzsMmjsD4rrdC6~EnLZONTEXKTSbrwNgk2Wh5WOWSdrOs09VwlQqxg0IAZqwEl6sgnYKGThYgTSn8N3fyVuhE1nkv4FGgcgJugYsquuHrUb6FvBVx01GammyHX9nvQYrydveGEXdz4B3aYSQkR9UC4UHIUGXejFvJtExQMf1kOmyj9floJrtnMIfFGyUxsAKiAuO1bPp1cPcGs8rv3fX88dDEQMLSoM52-c2kN5PeDj2XkfyWKdUbwiSktcFD4w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Age and CR rate of biCEBPACCSpos, biCEBPACCSneg, and moCEBPA patients. (A) Age of biCEBPACCSneg (median = 48), biCEBPACCSpos (median = 66), and moCEBPA (median = 62) patients. Scatter dot plot, median with interquartile range. (B) The CR rate was significantly higher in biCEBPACCSneg patients (91% [20/22]) than biCEBPACCSpos (61% [14/23]; P = .035) and moCEBPA patients (P = .027).
Age and CR rate of biCEBPACCSpos, biCEBPACCSneg, and moCEBPA patients. (A) Age of biCEBPACCSneg (median = 48), biCEBPACCSpos (median = 66), and moCEBPA (median = 62) patients. Scatter dot plot, median with interquartile range. (B) The CR rate was significantly higher in biCEBPACCSneg patients (91% [20/22]) than biCEBPACCSpos (61% [14/23]; P = .035) and moCEBPA patients (P = .027).
Characteristics of biCEBPACCSneg vs biCEBPACCSpos patients
| Characteristic | biCEBPACCSneg | biCEBPACCSpos | P |
|---|---|---|---|
| Age, y | .005 | ||
| Median | 48 | 66 | |
| Range | 20-71 | 28-84 | |
| Sex | .77 | ||
| Female | 12 (52) | 11 (44) | |
| WBC count, ×109/L | 24/25 | .39 | |
| Median | 11.8 | 30.8 | |
| Range | 1.3-408.6 | 1.7-284.4 | |
| Hemoglobin, g/dL | 21/23 | 23/25 | .12 |
| Median | 10.1 | 9.7 | |
| Range | 7.9-13.8 | 6.4-12.1 | |
| Platelet count, ×109/L | 24/25 | .89 | |
| Median | 28 | 35 | |
| Range | 3-151 | 10-120 | |
| Bone marrow blasts, % | 19/23 | 21/25 | .58 |
| Median | 66 | 75 | |
| Range | 20-97 | 31-100 | |
| LDH, U/dL | 19/23 | 17/25 | .14 |
| Median | 427 | 561 | |
| Range | 205-2510 | 229-1660 |
| Characteristic | biCEBPACCSneg | biCEBPACCSpos | P |
|---|---|---|---|
| Age, y | .005 | ||
| Median | 48 | 66 | |
| Range | 20-71 | 28-84 | |
| Sex | .77 | ||
| Female | 12 (52) | 11 (44) | |
| WBC count, ×109/L | 24/25 | .39 | |
| Median | 11.8 | 30.8 | |
| Range | 1.3-408.6 | 1.7-284.4 | |
| Hemoglobin, g/dL | 21/23 | 23/25 | .12 |
| Median | 10.1 | 9.7 | |
| Range | 7.9-13.8 | 6.4-12.1 | |
| Platelet count, ×109/L | 24/25 | .89 | |
| Median | 28 | 35 | |
| Range | 3-151 | 10-120 | |
| Bone marrow blasts, % | 19/23 | 21/25 | .58 |
| Median | 66 | 75 | |
| Range | 20-97 | 31-100 | |
| LDH, U/dL | 19/23 | 17/25 | .14 |
| Median | 427 | 561 | |
| Range | 205-2510 | 229-1660 |
Data represent number or n (%) of patients, unless otherwise indicated. Bold indicates significant P values.
biCEBPACCSpos patients had a trend to poorer outcome in terms of OS than biCEBPACCSneg patients (hazard ratio [HR], 2.6; 95% confidence interval [CI], 0.9-7.4; P = .078; Figure 4A). RFS and cumulative incidence of relapse were not statistically significant (supplemental Figure 4)
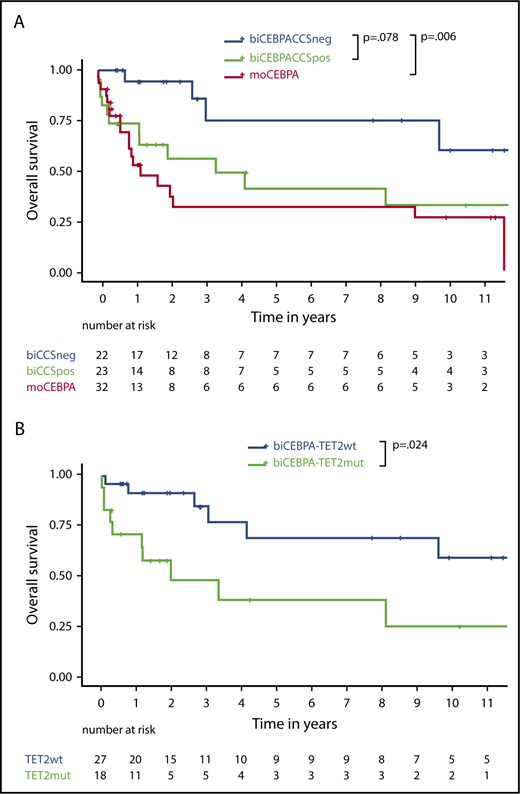
Survival data depending on biogroup and TET2 status. (A) OS of biCEBPACCSneg, biCEBPACCSpos, and moCEBPA patients. (B) OS of biCEBPA patients depending on TET2 mutational status.
Survival data depending on biogroup and TET2 status. (A) OS of biCEBPACCSneg, biCEBPACCSpos, and moCEBPA patients. (B) OS of biCEBPA patients depending on TET2 mutational status.
Multivariable Cox regression including CCS status and age suggested that age does not fully explain the potential differences in OS (adjusted CCS HR, 2.0; 95% CI, 0.7-6.0; P = .22; supplemental Table 5). In a multivariable logistic regression model to assess the association between CR and CCS after adjusting for age, the score was prognostic for patients in CR (adjusted CCS OR, 0.17; 95% CI, 0.03-1.01; P = .05), but not patients in CR or CR with incomplete hematologic recovery (adjusted CCS OR, 0.58; 95% CI, 0.08-3.97; P = .58; supplemental Table 7). For the end point early death at day 60, the model proved to be unsuitable, because there was no early-death event in the biCEBPACCSneg cohort and only 4 in the biCEBPACCSpos cohort (supplemental Table 6). Accounting for a reduced statistical power due to small numbers, CCS status might have an independent influence on survival. Survival of biCEBPACCSpos and moCEBPA patients was comparable (Figure 4A).28
biCEBPACCSpos and biCEBPACCSneg patients did not significantly differ in treatment regimens, which consisted of high-dose cytarabine and mitoxantrone vs 6-thioguanine, standard-dose cytarabine and daunorubicin (TAD9) (supplemental Table 8).
As most of the patients in biCEBPACCSpos group (20/25, 80%) were defined by TET2 mutations, we further analyzed the prognostic effect of TET2. OS (HR, 3.1; 95% CI, 1.2-8.1; P = .024) was significantly worse in biCEBPA patients with a TET2 mutation than those without a TET2 mutation (Figure 4B). In biCEBPA patients, there was a trend for an effect of TET2 on RFS (supplemental Figure 5).
We also analyzed OS and RFS in moCEBPA patients (supplemental Figure 6A-B). In 32 (n = 14 TET2 mutated) and 21 (n = 8 TET2 mutated) moCEBPA patients, there was no significant difference in OS (HR, 2.0; 95% CI, 0.8-5.2; P = .15) and RFS (HR, 0.9; 95% CI, 0.3-2.9; P = .99), respectively.
TET2 and DNTM3A mutations, which largely define our biCEBPACCSpos subgroup, are frequently present during complete morphologic remission as a result of clonal hematopoiesis.29,30 This finding leads us to the hypothesis that in some biCEBPACCSpos patients, a preleukemic clone might have been present (supplemental Figure 7). In 4 out of 5 patients for whom material from complete morphologic remission was available, we could identify persisting TET2 or DNMT3A mutations (Figure 5A; supplemental Table 9). It is very likely that in these patients, the TET2 and DNMT3A mutations might have preceded the CEBPA mutations. In contrast, we did not find persisting mutations in available remission samples (n = 5) from the biCEBPACCSneg subgroup (Figure 5B).
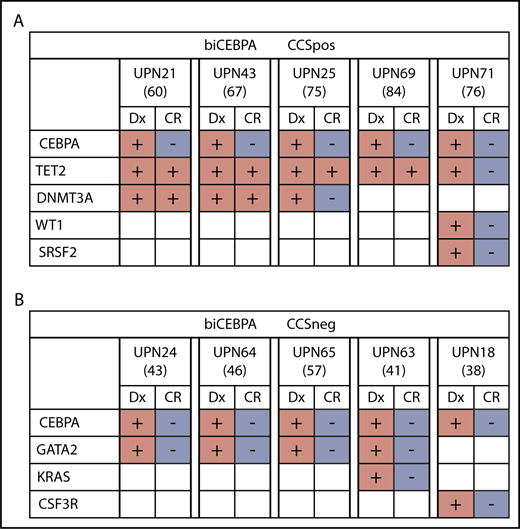
Mutational status of biCEBPACCSposand biCEBPACCSnegremission samples (age is given in brackets). (A) Four out of 5 biCEBPACCSpos patients had persisting clones in CR, indicating a clonal hematopoiesis. (B) None of the 5 biCEBPACCSneg samples had a detectable mutation load at the time of remission. Dx, diagnosis.
Mutational status of biCEBPACCSposand biCEBPACCSnegremission samples (age is given in brackets). (A) Four out of 5 biCEBPACCSpos patients had persisting clones in CR, indicating a clonal hematopoiesis. (B) None of the 5 biCEBPACCSneg samples had a detectable mutation load at the time of remission. Dx, diagnosis.
In 6% to 10% of biCEBPA-mutated AML patients, 1 of the CEBPA mutations is of germline origin.31,32 Because of younger age and fewer or no concomitant mutations, a germline CEBPA variant would be more likely to occur in biCEBPACCSneg patients. Remission material was available from 8 out of 23 biCEBPACCSneg samples, and these patients had no persisting CEBPA mutation. In 7 out of 25 biCEBPACCSpos samples, we also did not detect a CEBPA germline mutation.
Discussion
Our study provides novel key findings with genetic relevance in CEBPA-mutated AML. We studied a cohort of 80 CEBPA-mutated CN-AML patients, including 48 patients with biallelic mutations. Samples from these patients were genetically well characterized by deep amplicon sequencing, with a special focus on mutation patterns between moCEBPA and biCEBPA patient samples and within the biCEBPA subgroup.
In this dataset, we identified novel associations of moCEBPA with STAG2 and FLT3- TKD1/2 mutations.2,23 Previously, Lavallee et al identified recurrent mutations (T618I) in the CSF3R (29%) gene in a small cohort of 14 patients with double-mutated CEBPA.14 CSF3R is the receptor for colony-stimulating factor 3 and, like CEBPA, is crucial for normal granulopoiesis.33 In this cohort of biCEBPA patients, the mutation frequency of the T618I CSF3R was 10%. Because of the larger size of our biCEBPA cohort and the fact that we only included normal-karyotype-AML the mutation frequency of CSF3R in our cohort might be more representative of biCEBPA CN-AML patients.
We found a higher frequency of TET2 mutations in moCEBPA (44%) and biCEBPA patients (42%) than in other AML or CEBPA-mutated AML cohorts (8% to 24%).23-25 When we compared these data with our wtCEBPA cohort data set, we found that TET2 mutations define a distinct subgroup of CEBPA-mutated samples regardless of allelic status. Most mutations result in a frame shift or premature stop codon, underscoring their functional relevance. The presence of a TET2 mutation in biCEBPA patients has a negative impact on OS, which is in concordance with the findings of Grossmann et al.31
We also could show that patients in the biCEBPA subgroup had a significantly lower frequency of cooccurring mutations. Although we did not perform genome-wide analysis, these data suggest that the second hit in the CEBPA gene may drive leukemogenesis, and fewer cooperating mutations in other genes might therefore be required.
We identified 2 genetic subgroups in biCEBPA-mutated AML and validated this finding in a biCEBPA patient cohort from Papaemmanuil et al.21 Patients in the biCEBPACCSpos group were significantly older and had a poorer prognosis, and most patients with remission samples available had persisting mutations in TET2 and/or DNMT3A, suggesting preleukemic clonal hematopoiesis even before the onset of leukemia. These data suggest that CCS mutations are the initiating events in this subgroup, followed by the acquisition of biCEBPA mutations. In contrast, biCEBPACCSneg patients were younger, had a unique pattern of co-occurring mutations, cleared all mutations in remission, and had a favorable prognosis.
Two patients with moCEBPA mutation also had persisting DNMT3A mutations. Due to a lack of samples, we could not perform any further analysis in this cohort.
The poorer prognosis, higher early death rate, and lower CR rate in biCBEPACCSpos patients could only partly be explained by older age. Clonal hematopoiesis might also be a risk factor for early death, although our study lacks the statistical power to test this hypothesis. Recent data indicate that healthy individuals with clonal hematopoiesis of indeterminate potential have a higher risk of cardiovascular disease.34 In a logistic regression model, the CCS score for biCEBPA-mutated patient samples was prognostic for patients in CR.
In conclusion, our results suggest that biCEBPA mutations can be further subdivided by CCS mutation status in 2 groups with distinct genetic characteristics. This subclassification provides insight in the pathogenesis of the disease and might help to further refine prognostic classification of AML.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Gudrun Mellert, Evelyn Packeiser, and Kathrin Bräundl (Ludwig-Maximilians-Universität) as well as Oliver Wachter (Center for Human Genetics and Laboratory Medicine, Martinsried, Germany) for excellent technical support. The authors acknowledge the contribution of all participants of the AMLCG trial and recruiting centers.
AMLCG clinical trials were supported by Deutsche Krebshilfe. This study was supported by the Deutsche Forschungsgemeinschaft (DFG SFB 1243, TPA07) (K.S.). S.K.B. is supported by Leukaemia & Blood Cancer New Zealand and the family of Marijanna Kumerich.
Authorship
Contribution: N.P.K., A.D., and K.S. designed the study; N.P.K., K.H.M., and M.R.-T. analyzed sequencing data; N.P.K., E.H., F.P., and T. Herold performed statistical analysis; N.P.K., S.K.B., S.S., B.K., S.T., and A.D. were involved in the laboratory characterization of patient samples; F.P. and M.C.S. provided clinical follow-up data. T. Hinrichsen and H.-G.K. performed sequencing on the Ion PGM system and analyzed the data; J.B., W.E.B., B.J.W., M.C.S., and W.H. coordinated the AMLCG clinical trial; K.S., J.B., W.H., S.K.B., and K.H.M. were involved in patient care; N.P.K., K.S., F.P., and T. Herold wrote the manuscript with help from all authors; K.S. supervised the project; and all authors had access to primary clinical trial data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Karsten Spiekermann, Department of Internal Medicine III, University Hospital, LMU, Campus Grosshadern, Marchioninistr 15, 81377 Munich, Germany; e-mail: karsten.spiekermann@med.uni-muenchen.de.

