Key Points
B-cell deficiency and DNA damage in the bone marrow of Il10−/− mice are associated with inflammation and mitigated by antibiotics.
In ETV6-RUNX1+Cdkn2a−/− mice, low IL-10 accelerates the development of B-cell leukemia/lymphoma in a dose-dependent manner.

Abstract
Exposures to a wide repertoire of common childhood infections and strong inflammatory responses to those infections are associated with the risk of pediatric B-cell acute lymphoblastic leukemia (B-ALL) in opposing directions. Neonatal inflammatory markers are also related to risk by unknown mechanism(s). Here, we demonstrate that interleukin-10 (IL-10) deficiency, which is associated with childhood B-ALL, indirectly impairs B lymphopoiesis and increases B-cell DNA damage in association with a module of 6 proinflammatory/myeloid-associated cytokines (IL-1α, IL-6, IL-12p40, IL-13, macrophage inflammatory protein-1β/CCL4, and granulocyte colony-stimulating factor). Importantly, antibiotics attenuated inflammation and B-cell defects in preleukemic Cdkn2a−/−Il10−/− mice. In an ETV6-RUNX1+ (E6R1+) Cdkn2a−/− mouse model of B-ALL, decreased levels of IL-10 accelerated B-cell neoplasms in a dose-dependent manner and altered the mutational profile of these neoplasms. Our results illuminate a mechanism through which a low level of IL-10 can create a risk for leukemic transformation and support developing evidence that microbial dysbiosis contributes to pediatric B-ALL.
Introduction
The reported effects of early life infections on childhood B-cell acute lymphoblastic leukemia (B-ALL) are highly complex.1 Although prior studies have focused on the protective role of surrogates of infectious exposure, such as early day care attendance2,3 and contact with older siblings,4 more recent studies have highlighted a stimulatory role of infections within the first year of life.5,6 To develop strategies for the prevention of childhood acute lymphoblastic leukemia (ALL), there is a need to understand the underlying mechanism(s) of how infectious stimuli promote or suppress its development. Variation in the key host factors that regulate response to infection might also impact the risk of ALL.
The timing of exposure to an infectious pathogen is a hallmark of the prevailing “delayed infection” hypothesis, which proposes that a lack of common infections (vectors) in infancy can promote the emergence of childhood ALL by driving abnormal immune responses to infections contracted later in childhood.7 We have identified altered cytokine levels at birth as a risk factor for childhood ALL.8,9 These observations suggest that, in addition to the timing of early childhood infections, congenital defects in the host immune system may influence the development of childhood ALL.
Neonatal deficiency in interleukin-10 (IL-10), a key cytokine in mediating early microbial homeostasis10,11 and immune response to infection,12,13 is a strong predictor of childhood B-cell malignancies.14 Children born with low levels of IL-10 have a 25-fold increased risk for developing ALL, whereas children with inherited IL10R deficiency have a high risk for developing B-cell non-Hodgkin lymphoma.8,15 IL-10 is a potent anti-inflammatory cytokine that suppresses myeloid cell activation, migration, and cytokine production by triggering downstream JAK/STAT signaling pathways.16 Very early onset of severe inflammatory bowel disease (IBD) is observed in children born with IL-10 and IL-10R deficiency.17 Additionally, Il10−/− mice have been used as a model system for IBD and colitis-associated colon cancer, in which bacterial infection and tumor-promoting inflammation drive double-stranded DNA breaks in intestinal cells.18-20 Of importance for understanding disease susceptibility, different mouse strain backgrounds and the particular microbiota present in different mouse colonies influence the likelihood of Il10−/− animals developing IBD.21 Although the gut is the primary site of inflammation in IL-10–deficient humans and mice, gut dysbiosis and inflammation can have distinct genotoxic effects on peripheral blood lymphocytes22,23 that might contribute to hematological malignancies.24 We hypothesize that low IL-10 levels may be a source of excessive inflammation that drives B-cell mutagenesis during the development of childhood B-cell leukemia/lymphoma.
Mouse models have been paramount in confirming the ability of inflammatory stimuli, such as lipopolysaccharide and various infectious pathogens, to drive the acquisition of secondary driver mutations in preleukemic B cells and promote the development of leukemia/lymphoma.7,25-27 However, these models have yet to elucidate the role of host immune deficiencies in the infectious etiology of ALL. To investigate whether host immune and microbial dysfunction can contribute to the development of childhood ALL, we crossed Il10−/− mice with the robust ETV6-RUNX1+ (E6R1+) Cdkn2a−/− model of childhood B-ALL28 (previously referred to as the Cre/TA1 Cdkn2a−/− model). We report that IL-10 deficiency leads to increased DNA damage in bone marrow B cells, this increased damage is a result of microbial dysbiosis, and IL-10–deficient animals show decreased latency of B-cell neoplastic disease. Given the association between inflammation and microbial dysbiosis in Il10−/− mice,21 these data are compatible with such dysbiosis being a source of DNA damage that contributes to lymphoid transformation.
Methods
Mice
B6.129P2-Il10tm1Cgn/J mice (referred to in the text as Il10−/− mice; MGI: 1857199) were purchased from The Jackson Laboratory (stock no. 002251) and crossed for >10 generations onto the FVB/N strain background. Control FVB/N mice were bred in-house. E6R1+Cdkn2a−/− mice (previously referred to as Cre/TA1 Cdkn2a−/− mice28 ) were maintained on the FVB/N strain background. B6.129 Il10−/− mice were crossed to E6R1+Cdkn2a−/− mice on the FVB/N background for ≥4 generations. Male and female mice were age matched for each experiment. All experiments were performed following institutional review and approval by the University of California, San Francisco Institutional Animal Care and Use Committee.
Flow cytometry
Cells were stained with 4′,6-diamidino-2-phenylindole or Ghost Dye Violet 450 to measure viability by flow cytometry. After viability staining, cells were blocked in Fc Block or Rat IgG for 10 minutes in the dark on ice. Without washing, primary surface staining antibodies were added for an additional 20 minutes. Cells stained with biotinylated surface antibodies were then stained with fluorochrome-labeled streptavidin for 20 minutes. For intracellular staining, fixation and permeabilization were done using the BD Cytofix/Cytoperm Kit per the manufacturer’s instructions. Data were acquired using a Sony SP6800 Spectral Analyzer (Sony Biotechnology) and analyzed using FlowJo software.
Peripheral blood analyses
For peripheral blood plasma, whole blood was obtained through submandibular bleeding and collected in EDTA-coated tubes. For cytokine analysis, tubes were centrifuged for 10 minutes at 1600g at 4°C to remove blood cells. Cytokine analysis was run on 2 technical replicate plasma samples from each mouse. A total of 32 cytokines were measured using 9-plex [IL-15, IL-18, Basic FGF, LIF, macrophage colony-stimulating factor, MIG, macrophage inflammatory protein-2 (MIP-2), PDGF-BB, and vascular endothelial growth factor] and 23-plex [IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL12p40, IL-12p70, IL-13, IL-17A, eotaxin, granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor, interferon-γ, KC, monocyte chemoattractant protein-1, MIP-1α, MIP-1β, RANTES, and tumor necrosis factor-α (TNF-α)] Luminex bead-based assays (Bio-Rad Laboratories).
Antibiotic treatment
Nutritionally complete pellets containing amoxicillin, clarithromycin, metronidazole, and omeprazole (Bio-Serv) were provided to mice for experiments shown in Figure 3C-G.
Histology
At the time of euthanasia, a gross necropsy was performed to identify potential sources of illness, and selected tissues were placed into 10% buffered formalin. If gross necropsy findings suggested a hematopoietic neoplasm, single-cell suspensions of involved tissues were prepared when possible and cryopreserved in medium containing 10% dimethyl sulfoxide. Formalin-fixed paraffin embedded (FFPE) tissues were stained with hematoxylin and eosin. Diagnoses were based upon gross necropsy and histopathology. Additional diagnostic information was obtained by immunophenotyping when necessary.
DNA extraction
The DNeasy and QIAamp DNA FFPE Tissue kits (QIAGEN) were used to extract DNA from cryopreserved or FFPE samples from leukemic mice. DNA concentration and quality were determined by Nanodrop spectrophotometry and PicoGreen (Invitrogen).
Statistical analysis
Statistical analyses were performed using Prism 8 software (GraphPad). P values were generated using an unpaired 2-tailed Mann-Whitney U test or a paired 2-tailed Student t test for single comparisons. A χ2 test was used for multiple comparisons. Correlations were calculated using the Pearson correlation. Survival analyses were performed using the log-rank/Mantel-Cox test. Analysis of Cytomod data in Figure 2 used statistical tests that are previously described.29
See supplemental Methods for addition details on cell preparation, sequencing, and analysis of sequencing.
Results
B-cell lymphopoiesis is altered in Il10−/− mice
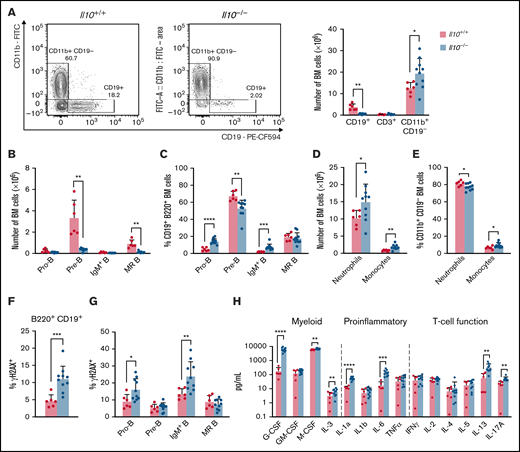
To investigate the impact of IL-10 deficiency on hematopoietic development, we enumerated lymphoid and myeloid lineages in the bone marrow of 8- to 12-week-old FVB/N Il10-knockout mice. (These mice were Il10−/−Cdkn2a+/+; see sections starting at "IL-10 deficiency disrupts B-cell properties in preleukemic Cdkn2a−/− mice" regarding Il10−/−Cdkn2a−/− mice.) Within the bone marrow of Il10−/− mice we observed decreased CD19+ B cells, increased CD11b+ CD19− myeloid cells, and no change in CD3+ T cells (Figure 1A). The substantial loss of bone marrow B cells was consistent with Il10−/− mice on the 129Sv/Ev background.30 A well-documented cause of bone marrow B-cell lymphopenia is the premature mobilization of pre-B cells to the periphery in response to inflammation and increased neutrophil production.31 Analysis of the CD19+B220+ B-cell compartment showed a reduction in the number of pre-B cells (CD43−IgD−IgM−) and mature recirculating (MR) B cells (IgD+IgM+), whereas the number of pro-B cells (CD43+IgD−IgM−) and IgM+ immature B cells (IgM+IgD−) remained unchanged (Figure 1B). As a result of the drastic reduction in pre-B cells, pro-B cells and IgM+ B cells were increased as a percentage of CD19+B220+ bone marrow B cells (Figure 1C). Neutrophil and monocyte numbers were increased in Il10−/− bone marrow (Figure 1D); monocytes were increased as a percentage of these CD11b+CD19− cells (Figure 1E). B-cell loss in the bone marrow was associated with changes in the spleen, including splenomegaly, increased cellularity, and expansion of mature myeloid cells, but it was not attributed to the mobilization of B cells to the spleen (supplemental Figure 1A-C). Together, these data show that a deficit of pre-B cells and MR B cells occurs concurrently with myeloid cell expansion in Il10−/− mice.
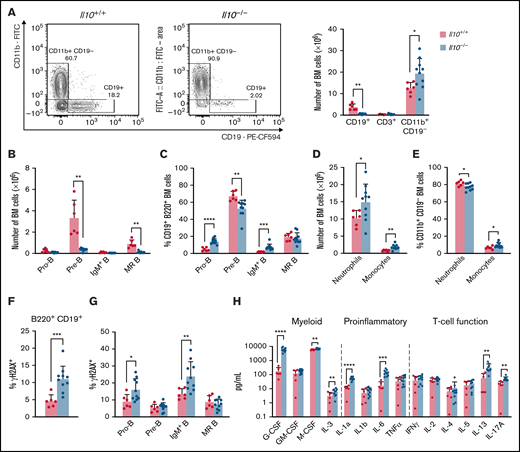
Disruption of B lymphopoiesis and double-stranded DNA breaks in B cells are correlated with myeloid expansion in Il10−/− mice. Analysis of immune cell lineages and cytokines in 8- to 12-week-old Il10+/+ and Il10−/− mice. (A) Representative flow cytometry plots show the percentage of CD11b+ CD19− myeloid cells and CD19+ B cells of FVB/N and Il10−/− bone marrow (BM) gated on single live cells (left panel). Bar graph summarizes the number of CD19+ B, CD3+ T, and CD11b+ CD19− myeloid cells in the BM (right panel). (B) Number of pro-B cells (B220+CD19+IgM−IgD−CD43+), pre-B cells (B220+CD19+IgM−IgD−CD43−), IgM+ immature B cells (B220+CD19+IgM+IgD−), and MR B cells (B220+CD19+IgM+IgD+) in the BM. (C) Percentage of B-cell subsets in B220+CD19+ BM. (D) Number of neutrophils (CD11b+CD19− Gr1hiLy6Clo) and monocytes (CD11b+CD19−Gr1loLy6Chi) in the BM. (E) Percentage of myeloid subsets in single live BM cells. Percentage of γH2AX+ cells among total BM B cells (F) and BM B-cell subsets (G). (H) Absolute plasma concentrations of a selected panel of myeloid, proinflammatory, and T-cell regulatory cytokines detected by a bead-based multiplex Luminex assay. Plasmas of all mice were assayed simultaneously. Nominal P values are presented for cytokine data. Data in (A-G) are representative of 3 experiments. Error bars represent mean ± standard deviation. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .001. Statistical tests used are described in Methods.
Disruption of B lymphopoiesis and double-stranded DNA breaks in B cells are correlated with myeloid expansion in Il10−/− mice. Analysis of immune cell lineages and cytokines in 8- to 12-week-old Il10+/+ and Il10−/− mice. (A) Representative flow cytometry plots show the percentage of CD11b+ CD19− myeloid cells and CD19+ B cells of FVB/N and Il10−/− bone marrow (BM) gated on single live cells (left panel). Bar graph summarizes the number of CD19+ B, CD3+ T, and CD11b+ CD19− myeloid cells in the BM (right panel). (B) Number of pro-B cells (B220+CD19+IgM−IgD−CD43+), pre-B cells (B220+CD19+IgM−IgD−CD43−), IgM+ immature B cells (B220+CD19+IgM+IgD−), and MR B cells (B220+CD19+IgM+IgD+) in the BM. (C) Percentage of B-cell subsets in B220+CD19+ BM. (D) Number of neutrophils (CD11b+CD19− Gr1hiLy6Clo) and monocytes (CD11b+CD19−Gr1loLy6Chi) in the BM. (E) Percentage of myeloid subsets in single live BM cells. Percentage of γH2AX+ cells among total BM B cells (F) and BM B-cell subsets (G). (H) Absolute plasma concentrations of a selected panel of myeloid, proinflammatory, and T-cell regulatory cytokines detected by a bead-based multiplex Luminex assay. Plasmas of all mice were assayed simultaneously. Nominal P values are presented for cytokine data. Data in (A-G) are representative of 3 experiments. Error bars represent mean ± standard deviation. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .001. Statistical tests used are described in Methods.
Other investigators have reported that early hematopoietic progenitors in Il10−/− mice have a decreased capacity for sustaining long-term culture.32 We observed that the bone marrow of young 4- to 6-week-old Il10−/− mice had decreased numbers of common myeloid progenitor, granulocyte/monocyte progenitor, and megakaryocyte/erythrocyte progenitor cells, with no change observed in Lin−Sca-1+c-kit+ or common lymphoid progenitor cells (supplemental Figure 1D). No difference in hematopoietic stem and progenitor cell number was observed in the spleen of young Il10−/− mice (supplemental Figure 1E).
IL-10 deficiency contributes to B-cell DNA damage
The pathogenesis of pediatric B-ALL in humans and mice is associated with RAG-mediated double-stranded DNA breaks in immunoglobulin and nonimmunoglobulin genes.25,33 Furthermore, other investigators have reported increased double-stranded DNA breaks in peripheral blood lymphocytes and small intestinal cells of Il10−/− mice.20 To investigate whether IL-10 deficiency may contribute to bone marrow B-cell transformation, we used flow cytometry to measure γH2AX, a DNA repair protein and double-stranded DNA break marker, in B-cell progenitors from 8- to 12-week-old Il10−/− mice. The percentage of γH2AX+ B cells was significantly increased in Il10−/− mice relative to Il10+/+ controls (Figure 1F). DNA damage in Il10−/− B cells was most abundant in the pro-B cell and IgM+ immature B-cell subsets (Figure 1G). Overall, our results provide evidence that IL-10 deficiency negatively selects against pre-B cells and that the few remaining bone marrow pro-B cells and IgM+ immature B cells acquire DNA damage.
Inflammation in Il10−/− mice is associated with B-cell DNA damage
Inflammation is a well-established driver of DNA damage in human cancer34 and Il10−/− mouse models of cancer.35 We hypothesized that the expression of proinflammatory cytokines in the plasma of Il10−/− mice would be associated with the level of DNA damage in developing B cells. To test this hypothesis, we measured the concentration of 32 cytokines that are involved in T helper 1 (Th1) cell, Th2 cell, Th17 cell, and innate immune responses. The following 6 cytokines were significantly increased in Il10−/− mice: IL-1α, IL-6, IL-12p40, IL-17, G-CSF, and MIP-1β (CCL4) (corrected P value < .05; Table 1). Although previous work reported a bias toward the expression of the Th1 cell–related cytokines interferon-γ and TNF-α in Il10−/− mice,36 we demonstrate that myeloid growth factors and proinflammatory cytokines can predominate the peripheral blood inflammatory response in Il10−/− mice (Figure 1H).
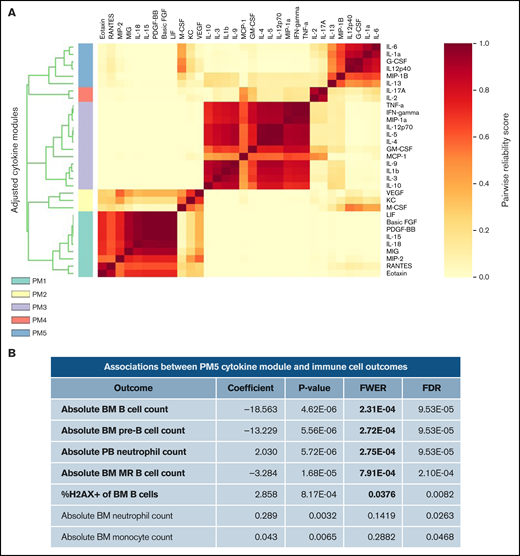
We then tested whether patterns of inflammation were associated with abnormal B-cell numbers and DNA damage in Il10−/− mice. During complex immune responses, such as inflammation, cytokines may change in characteristic patterns. Therefore, we applied clustering analysis to our cytokine data set to determine whether cytokine clusters were more significantly associated with B-cell DNA damage and proliferation compared with individual cytokines. To this end, we used CytoMod,29 an unsupervised approach to clustering cytokines across individuals. We then used linear regression to determine whether there was an association between cytokine modules and altered B-cell number and DNA damage. P values were adjusted using the Bonferroni-Holm method to obtain the family-wise error rate (FWER). We chose FWER < 0.05 as a cutoff for statistical significance. Of the 5 cytokine modules that were derived from normalized cytokine concentrations (Figure 2A), plasma module 5 (PM5) was associated with an increased percentage of DNA damage in bone marrow B cells and decreased numbers of pre-B and MR B cells (Figure 2B). PM5 contained the following cytokines: IL-1α, IL-6, IL-12p40, IL-13, G-CSF, and MIP-1β (Figure 2A). Our results demonstrate that, in the setting of IL-10 deficiency, proinflammatory cytokines are associated with B-cell loss and DNA damage. These results also indicate that IL-10 deficiency can contribute to the aberrant expression of the proinflammatory cytokine IL-6, which is a causal factor for childhood B-ALL, as proposed previously in neonatal blood9 and demonstrated more recently in mice.37
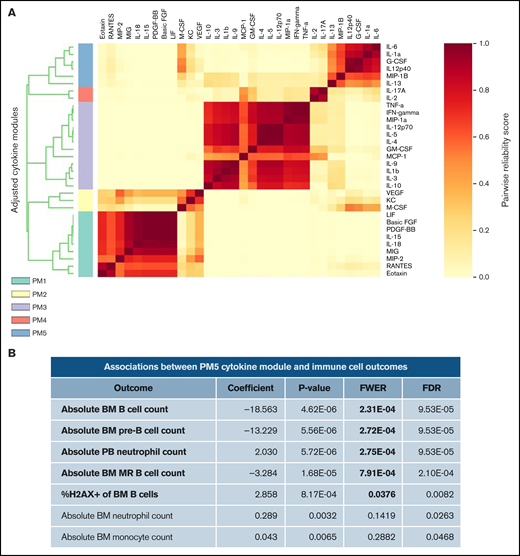
Cytokine modules defined in combined cytokine profiles of Il10+/+ and Il10−/− mice are associated with B-cell outcomes. (A) Heat map of cytokine modules displaying pairwise reliability scores of adjusted cytokines over 1000 random samplings of subjects. The optimal number of clusters was determined by the Tibshirani gap statistic method. (B) Table of associations between PM5 module and immune cell outcomes with differential abundance between Il10+/+ and Il10−/− bone marrow (BM) or peripheral blood (PB). Log-cytokine regression coefficient, P value, FWER, and false-discovery rate (FDR) were calculated as described.29
Cytokine modules defined in combined cytokine profiles of Il10+/+ and Il10−/− mice are associated with B-cell outcomes. (A) Heat map of cytokine modules displaying pairwise reliability scores of adjusted cytokines over 1000 random samplings of subjects. The optimal number of clusters was determined by the Tibshirani gap statistic method. (B) Table of associations between PM5 module and immune cell outcomes with differential abundance between Il10+/+ and Il10−/− bone marrow (BM) or peripheral blood (PB). Log-cytokine regression coefficient, P value, FWER, and false-discovery rate (FDR) were calculated as described.29
IL-10 deficiency disrupts B-cell properties in preleukemic Cdkn2a−/− mice
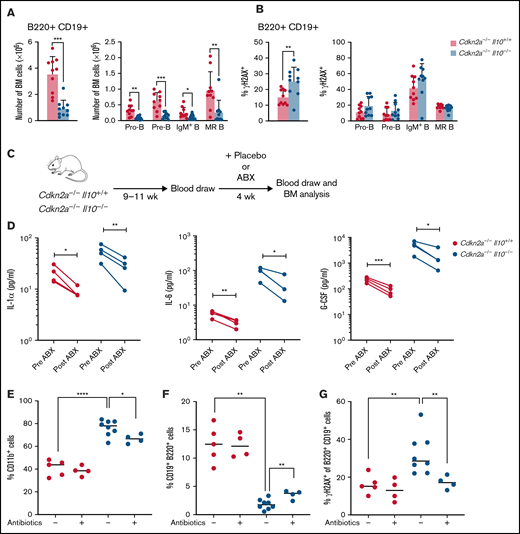
We then investigated the role of IL-10 deficiency in the Cdkn2a−/− mouse model of B-cell leukemia/lymphoma. For preleukemic studies, Cdkn2a−/− mice were Il10+/+ or Il10−/− (referred to herein as Cdkn2a−/−Il10+/+ or Cdkn2a−/−Il10−/−). Cdkn2a codes for the p16Ink4a and p19Arf (p14ARF in humans) tumor suppressor proteins, which are key regulators of the cell cycle. CDKN2A loss is found in one third of cases of human ALL,38 and deletion of this gene in mice can induce B-ALL.39 Deletion of Il10 in Cdkn2a−/− mice had a similar effect on inducing pre-B-cell loss and B-cell DNA damage as did Il10 deletion in wild-type Cdkn2a mice (Figure 3A). However, we did not detect a particular B-cell subset with increased DNA damage (Figure 3B). Although age-matched Il10+/+ and Il10−/− mice were not assessed side by side with Cdkn2a−/−Il10+/+ and Cdkn2a−/−Il10−/− mice, it is worth noting that the fold change between Il10−/− and control mouse B-cell numbers (Figure 1A vs Figure 3A) and total B-cell DNA damage (Figure 1F vs Figure 3B) was similar across these different experiments. In addition, Cdkn2a−/−Il10−/− mice shared patterns of inflammation with Il10−/− mice: 15 cytokines were statistically significantly increased in Cdkn2a−/−Il10−/− mice (Table 2), including the 6 cytokines that were increased in Il10−/− mice. Therefore, the effects of IL-10 deficiency on B-cell lymphopoiesis and inflammation were present in the Cdkn2a−/− mouse model of B-cell leukemia/lymphoma.
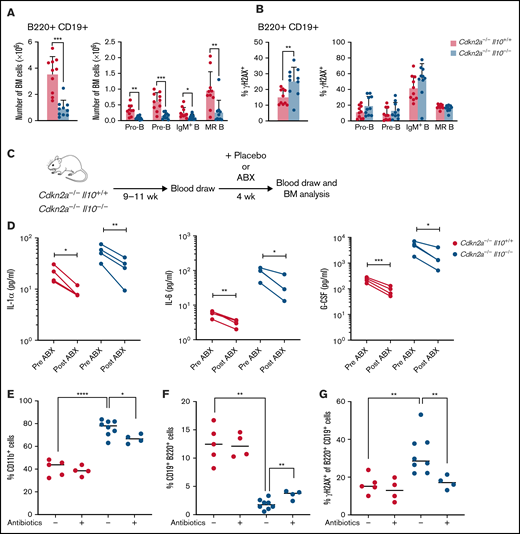
Antibiotic treatment response rescues preleukemic Cdkn2a−/−Il10−/− B cells from impaired development and DNA damage. Analysis of 8- to 12-week-old Cdkn2a−/−Il10+/+ and Cdkn2a−/−Il10−/− mice. (A) Absolute bone marrow (BM) count of total B220+CD19+ B cells and BM B-cell subsets. (B) Percent of γH2AX+ cells among total BM B cells and BM B-cell subsets. (C) Schematic diagram of antibiotic (ABX) treatment and tracking of peripheral blood and BM cells in adult mice. (D) Concentration of cytokines in Il10−/−Cdkn2a−/− mice and controls. Lines connect values from the same mouse sampled before and after antibiotic treatment. Flow analysis of the percentage of CD11b+CD19− cells (E) and CD19+ B220+ cells (F) in BM. (G) Percentage of γH2AX+ of CD19+B220+ cells in Cdkn2a−/−Il10+/+ and Cdkn2a−/−Il10−/− mice after treatment with placebo (-) or antibiotics (+) for 4 weeks. Data in (A-B) are representative of 2 experiments. Data in (D-G) were obtained from a single-cohort study. Error bars represent the mean ± standard deviation. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .001. Statistical tests used are described in Methods.
Antibiotic treatment response rescues preleukemic Cdkn2a−/−Il10−/− B cells from impaired development and DNA damage. Analysis of 8- to 12-week-old Cdkn2a−/−Il10+/+ and Cdkn2a−/−Il10−/− mice. (A) Absolute bone marrow (BM) count of total B220+CD19+ B cells and BM B-cell subsets. (B) Percent of γH2AX+ cells among total BM B cells and BM B-cell subsets. (C) Schematic diagram of antibiotic (ABX) treatment and tracking of peripheral blood and BM cells in adult mice. (D) Concentration of cytokines in Il10−/−Cdkn2a−/− mice and controls. Lines connect values from the same mouse sampled before and after antibiotic treatment. Flow analysis of the percentage of CD11b+CD19− cells (E) and CD19+ B220+ cells (F) in BM. (G) Percentage of γH2AX+ of CD19+B220+ cells in Cdkn2a−/−Il10+/+ and Cdkn2a−/−Il10−/− mice after treatment with placebo (-) or antibiotics (+) for 4 weeks. Data in (A-B) are representative of 2 experiments. Data in (D-G) were obtained from a single-cohort study. Error bars represent the mean ± standard deviation. *P ≤ .05, **P ≤ .01, ***P ≤ .001, ****P ≤ .001. Statistical tests used are described in Methods.
We observed variable penetrance of inflammation, as defined by excessive proinflammatory cytokines and corresponding increased monocytes, in Il10−/− and Cdkn2a−/−Il10−/− mice. To assess the potential for inflammation to drive B-cell abnormalities, 8- to 12-week-old preleukemic Cdkn2a−/−Il10−/− mice were stratified into 2 groups based on the presence or absence of increased inflammatory cells relative to control Cdkn2a−/−Il10+/+ mice. A comparison of Cdkn2a−/−Il10−/− mice with normal myeloid counts with those with elevated monocytes or elevated neutrophils (supplemental Figure 2A) revealed that abnormalities in B-cell count and DNA damage were only present in Cdkn2a−/−Il10−/− mice with elevated monocytes (supplemental Figure 2B-C). These results indicate that IL-10 deficiency depletes and damages bone marrow B cells in a manner that might rely on inflammatory monocytes. These results suggest that inflammatory monocytes in Cdkn2a−/−Il10−/− mice could be responsible for abnormalities in developing B cells, as previously reported for splenic marginal zone B cells.21
Antibiotic-mediated suppression of the inflammatory milieu promotes recovery of B-cell development and diminishes B-cell DNA damage in Il10−/− mice
Mice housed in specific pathogen–free facilities commonly harbor Helicobacter spp. In Il10−/− mice, microbial dysbiosis of Helicobacter can spontaneously induce colitis40 and lead to an expansion of splenic inflammatory monocytes/macrophages and marginal zone B cells.21 We reasoned that such previously described inflammatory responses to microbial dysbiosis may also be an underlying cause of B-cell deficiency and elevated B-cell DNA damage in the bone marrow of Il10−/− mice.
We investigated whether blocking microbial-driven inflammation could limit abnormalities in B-cell number and DNA damage in preleukemic mice. To this end, we administered a 4-week regimen of antibiotics that target Helicobacter spp. or a placebo treatment to Cdkn2a−/−Il10+/+ and Cdkn2a−/−Il10−/− mice (Figure 3C). A multiplex Luminex assay was used to monitor cytokine concentrations in the peripheral blood before and after antibiotic treatment. Strikingly, antibiotic treatment reduced several proinflammatory cytokines that were associated with B-cell deficiency and B-cell DNA damage, including IL-1α, IL-6, and G-CSF (Figure 3D). We next assessed the impact of antibiotics on the frequency of immune cells and the extent of DNA damage in B cells residing in the bone marrow. As expected, the expansion of myeloid cells within the bone marrow compartment of Cdkn2a−/−Il10−/− mice was attenuated by antibiotics (Figure 3E). Antibiotic treatment also partially abrogated the decrease in B-cell number (Figure 3F) and remarkably reversed the increased B-cell DNA damage (Figure 3G) that was observed in placebo-treated Cdkn2a−/−Il10−/− mice. These results support a role for microbial dysbiosis and inflammation in promoting B-cell loss and B-cell DNA damage in the bone marrow of preleukemic Il10−/− mice.
Decreased levels of IL-10 accelerate B-cell neoplasms in E6R1 Cdkn2a−/− mice
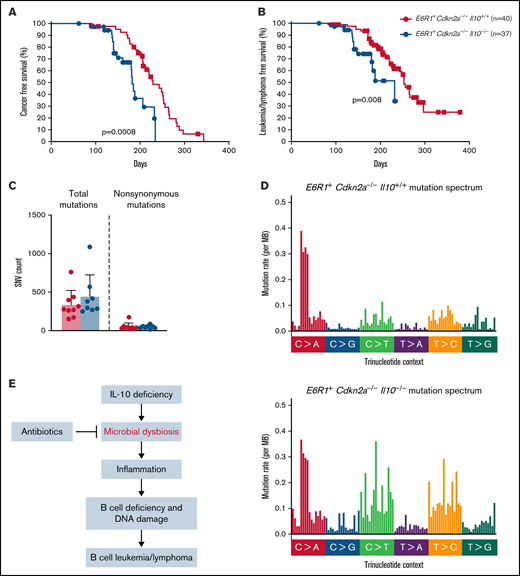
Children born with low levels of IL-10 were found to have a 25-fold increased risk for developing ALL.8 Whether this association reflects causality had not been previously tested. Although Cdkn2a−/− mice develop B-cell leukemia/lymphoma, a greater proportion of Cdkn2a−/− mice develop this malignancy in the presence of E6R1. Therefore, E6R1+Cdkn2a−/− mice were used to determine whether low levels of IL-10 promote the development of B-cell leukemia/lymphoma. We followed a cohort of E6R1+Cdkn2a−/− mice with wild-type IL-10 (E6R1+Cdkn2a−/−Il10+/+) for the development of disease alongside cohorts of mice with decreased IL-10 expression (E6R1+Cdkn2a−/−Il10+/−) or no IL-10 expression (E6R1+Cdkn2a−/−Il10−/−). We did not observe a significant difference in the incidence of leukemia/lymphomas among our survival cohorts (supplemental Table 1). Nevertheless, time-to-illness analysis revealed that a complete loss of IL-10 in E6R1+Cdkn2a−/−Il10−/− mice significantly decreased cancer-free survival (Figure 4A) and leukemia/lymphoma-free survival (Figure 4B) relative to E6R1+Cdkn2a−/−Il10+/+ controls. The median time to leukemia/lymphoma was 140 days in E6R1+Cdkn2a−/−Il10−/− mice vs 220 days in E6R1+Cdkn2a−/−Il10+/+ mice (P = 6.54 × 10−5, Mann-Whitney U test; supplemental Table 2). For E6R1+Cdkn2a−/−Il10+/− mice, the median time to leukemia/lymphoma was intermediate at 174 days (E6R1+Cdkn2a−/−Il10+/− vs E6R1+Cdkn2a−/−Il10+/+ mice, P = .01, Mann-Whitney U test; supplemental Table 2), supporting a dose-dependent relationship between decreased levels of IL-10 and early onset of leukemia/lymphoma (supplemental Figure 3). Of note, we were not able to subtype disease in all mice; however, consistent with previous work,28 B-cell leukemia/lymphoma was diagnosed in the preponderance of animals identified as having leukemia/lymphoma. These data demonstrate that low levels of IL-10 have a causal role in decreasing the time to leukemia/lymphoma development in the E6R1+Cdkn2a−/− model.
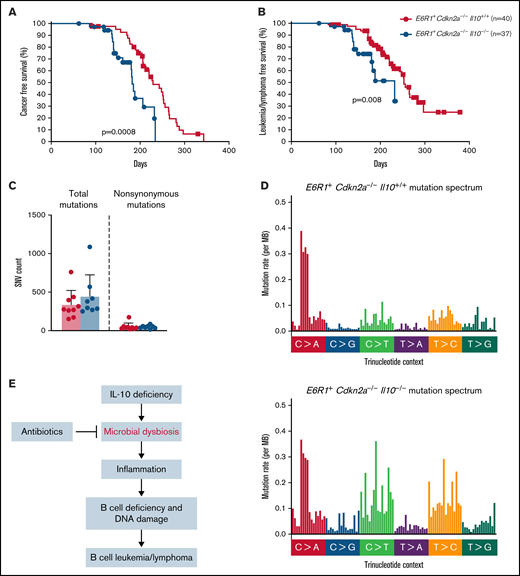
Decreased levels of IL-10 accelerate development of B-cell disease in the E6R1+Cdkn2a−/− model. Survival curves for cancer (A) and leukemia/lymphoma development (B) in E6R1+Cdkn2a−/−Il10+/+ mice and E6R1+Cdkn2a−/−Il10−/− mice. Log-rank (Mantel-Cox) test. (C) Number of total or nonsynonymous SNVs in B-cell leukemia/lymphomas from exome sequencing of 9 E6R1+Cdkn2a−/−Il10+/+ mice and 8 E6R1+Cdkn2a−/−Il10−/− mice. Error bars represent the mean ± standard deviation. (D) Mutation spectrum representing the frequency of mutations in each context of 96 possible trinucleotide contexts in sequenced E6R1+Cdkn2a−/−Il10+/+ and E6R1+Cdkn2a−/−Il10−/− B-cell leukemia/lymphomas. (E) Model for the role of microbial dysbiosis in childhood B-cell leukemia/lymphoma. IL-10 deficiency induces microbial dysbiosis in the gut, resulting in inflammation with distal effects of B-cell deficiency and B-cell DNA damage in the bone marrow. The inflammation-associated acquisition of genetic lesions in bone marrow B cells leads to the development of B-cell leukemia/lymphoma. Antibiotics may counteract the impact of low IL-10 by restoring bacterial homeostasis in the gut.
Decreased levels of IL-10 accelerate development of B-cell disease in the E6R1+Cdkn2a−/− model. Survival curves for cancer (A) and leukemia/lymphoma development (B) in E6R1+Cdkn2a−/−Il10+/+ mice and E6R1+Cdkn2a−/−Il10−/− mice. Log-rank (Mantel-Cox) test. (C) Number of total or nonsynonymous SNVs in B-cell leukemia/lymphomas from exome sequencing of 9 E6R1+Cdkn2a−/−Il10+/+ mice and 8 E6R1+Cdkn2a−/−Il10−/− mice. Error bars represent the mean ± standard deviation. (D) Mutation spectrum representing the frequency of mutations in each context of 96 possible trinucleotide contexts in sequenced E6R1+Cdkn2a−/−Il10+/+ and E6R1+Cdkn2a−/−Il10−/− B-cell leukemia/lymphomas. (E) Model for the role of microbial dysbiosis in childhood B-cell leukemia/lymphoma. IL-10 deficiency induces microbial dysbiosis in the gut, resulting in inflammation with distal effects of B-cell deficiency and B-cell DNA damage in the bone marrow. The inflammation-associated acquisition of genetic lesions in bone marrow B cells leads to the development of B-cell leukemia/lymphoma. Antibiotics may counteract the impact of low IL-10 by restoring bacterial homeostasis in the gut.
IL-10 loss increases the frequency of C>G and C>T mutations in B-cell neoplasms
We next sought to determine whether the increased levels of B-cell lesions observed in preleukemic Il10−/− mice corresponded to an increased mutational burden or altered mutational pattern in Il10−/− B-cell leukemia/lymphomas. We used whole-exome sequencing to characterize the number and type of mutations in B-cell leukemia/lymphomas from 9 E6R1+Cdkn2a−/−Il10+/+ mice and 8 E6R1+Cdkn2a−/−Il10−/− mice. Total and nonsynonymous single-nucleotide variants (SNVs) were similar in number between E6R1+Cdkn2a−/−Il10+/+ and E6R1+Cdkn2a−/−Il10−/− B-cell leukemia/lymphomas (Figure 4C). The total number of C>G and C>T mutations was increased in E6R1+Cdkn2a−/−Il10−/− samples (Figure 4D), although we did not find evidence for enrichment of inflammation-related mutational signatures delineated in the COSMIC mutational signatures database (version 2). Notably, C>T mutations are also elevated in solid tumors from mice with IL-10 deletions.41 SNVs related to human cancer were found in a number of genes (supplemental Table 3), with synonymous Hoxa9 mutations in 63% (5/8) of E6R1+Cdkn2a−/−Il10−/− B-cell leukemia/lymphomas. Similar synonymous mutations in HOXA9 have been found in several lymphoid neoplasm cell lines with a high pathogenicity score.42 Although synonymous changes have been considered nonpathogenic in the past, recent data suggest that such mutations have the potential to cause pathogenic effects.43 We also observed mutations in Jak3, Cdkn1a, and Dnm2 (supplemental Table 3), which have human orthologs that are mutated in human leukemias.44-47 The Jak3V670A mutation was present in 3 of 17 B-cell leukemia/lymphomas and is a homolog of human JAK3V674A. Mirroring the heterogeneity of human leukemias, other canonical cancer SNVs were identified in our cohort; however, there was little overlap in SNVs among samples. Overall, our results demonstrate that decreased levels of IL-10 cause acceleration of lymphoid malignancies with increased C>G and C>T mutations in the E6R1+Cdkn2a−/− model of B-cell leukemia/lymphoma.
Discussion
The prevailing delayed infection hypothesis regarding the infectious etiology of childhood leukemia posits that a lack of exposure to common pathogens during the first years of life raises the risk of leukemia.48 There is extensive support for a role of aberrant immune responses to common childhood infections in promoting leukemia development.49 However, the hypothesis that congenital defects in the host immune response can modulate the immune response to childhood infections, promoting the risk of childhood leukemia,50 has gained support by more recent studies.8,9,51 We observed that decreased levels of IL-10 caused a dose-dependent acceleration in the development of B-cell neoplasms in the E6R1+Cdkn2a−/− model of ALL. This finding provides in vivo evidence for a causal role of IL-10 deficiency in the development of B-cell neoplasms, building upon previous epidemiological studies that showed that low neonatal IL-10 is an ALL risk factor8 and that congenital genetic deficiencies in IL10 and IL10R are correlated with childhood B-cell lymphoma.15
Our data in preleukemic mice point to an indirect relationship between IL-10 deficiency and the impairment of B-cell development. In cohorts of Il10−/− mice with spontaneous onset of inflammation, none of the Il10−/− mice with normal monocyte counts (associated with a lack of inflammation) developed a deficit or extensive DNA damage within the bone marrow B-cell compartment. A role for inflammation in B-ALL development has been supported by several studies demonstrating that proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, can drive in vitro mutagenesis,37 selection of preexisting oncogenic mutations in B-cell progenitors,52 and in vivo B-ALL development in Pax5+/− mice.53 Given that IL-10 deficiency leads to the upregulation of a 6-cytokine module that is associated with B-cell DNA damage, chronic inflammation triggered by IL-10 deficiency may foster a favorable environment for B-cell transformation and leukemogenesis.
An additional mechanism for B-cell transformation and leukemogenesis might involve microbial-dependent inflammation caused by enterocolitis in Il10−/− mice. Gut bacteria in mice can drive VDJ recombination in lamina propria B cells54 and regulate hematopoiesis in the bone marrow.55 The attenuation of inflammation and B-cell DNA damage in antibiotic-treated Il10−/− mice indicates that microbial dysbiosis might underlie the genotoxic effects observed in bone marrow B cells. A different role of microbes, in protecting against childhood ALL, has been supported by human studies showing an association between newborns delivered through scheduled elective cesarean section and increased childhood ALL risk.56,57 In view of these human data, a study of ALL mouse models with intact IL-10 found that antibiotics increased leukemogenesis.58 The commonality between our findings and past studies is that childhood ALL development might be related to various states of microbial dysbiosis caused by excessive bacterial growth or deprivation of early microbial exposures. This is consistent with a concept, recently presented by Mel Greaves,49 that a well-balanced microbiome in early-life that promotes immunologic homeostasis may prevent childhood ALL.
Our work has limitations. We observed that low IL-10 levels accelerated development of B-cell neoplasms using a mouse model in which a combination of E6R1 expression with Cdkn2a deletion results in a relatively high incidence of leukemia/lymphoma.27 This penetrant development of disease may have limited our ability to detect the impact of IL-10 levels on disease incidence. In our model, large numbers of cells are susceptible to transformation (ie, initiated); hence, acceleration of disease may reflect low IL-10 levels increasing the probability of transformation (ie, progression). Nonetheless, studies of the impact of IL-10 levels in a low-penetrance model of lymphoblastic leukemia will be an important complement to our work. In addition, we do not yet have sufficient data to estimate the frequency at which low IL-10 levels are likely to be a contributory factor to cases of human pediatric leukemia/lymphoma. Further, although our mouse model system enabled us to identify that low IL-10 levels can lead to increased B-cell DNA damage and to an altered mutational spectrum, DNA damage studies of preleukemic cells, as well as sequencing of human ALLs from children with well-characterized cytokine profiles, are needed to assess whether similar phenomena occur in human leukemogenesis. With regard to the possibility of leukemia prevention, we showed that antibiotics can suppress B-cell DNA damage but did not demonstrate that such treatment could reverse leukemia acceleration.
Future prospective studies that are designed to characterize the development of the gut microbiome and immune system in children predisposed to ALL could further clarify the complex relationship among genetics, immune disorders, and microbial imbalances to ultimately guide clinical interventions aimed at decreasing ALL risk in children.
Acknowledgments
The authors thank Sony Biotechnology for providing guidance on the design of antibody staining panels for spectral flow cytometry and use of the Sony SP6800 Spectral Analyzer. They also thank Todd Whitehead and Kamir Hiam for useful discussions.
This work was supported by National Institutes of Health National Cancer Institute grants R01-CA185058 (S.C.K. and J.L.W.) and F31-CA221157 (B.A.F.).
Authorship
Contribution: B.A.F., M.L.H., J.L.W., and S.C.K. conceived the study; B.A.F., M.Z., J.S., S.S., M.Q.R., M.L.H., and S.C.K. designed the study; B.A.F., M.Z., J.S., and S.S. performed experiments and analyzed data; and B.A.F. wrote the manuscript with contributions by S.C.K. and M.Z. All authors interpreted data, reviewed the work critically, and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Scott C. Kogan, Helen Diller Family Comprehensive Cancer Center and Department of Laboratory Medicine, University of California San Francisco, 513 Parnassus Ave, Room S-561, Box 0451, San Francisco, CA 94143-0451; e-mail: scott.kogan@ucsf.edu.