Key Points
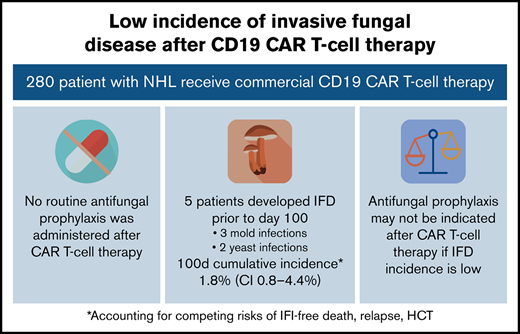
Among 280 patients receiving CD19 CAR T-cell therapy for NHL, the incidence of IFD was low despite no antifungal prophylaxis.
Antifungal prophylaxis can lead to toxicities or antifungal resistance and may not be indicated in institutions with low incidence of IFD.

Abstract
CAR T-cell therapy has revolutionized the treatment of hematologic malignancies, although its use may be complicated by toxicities, including cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), and infections. Invasive fungal disease (IFD) has been reported after CAR T-cell therapy, but the incidence in the absence of antifungal prophylaxis is unknown. Optimal prophylaxis strategies are widely debated. We performed a single-center retrospective study of 280 adults receiving CD19 CAR T-cell therapy for non-Hodgkin lymphoma (NHL) from December 2017 through September 2021. Patients did not receive routine antiyeast or antimold prophylaxis. IFD was identified between day of cell infusion and last follow-up. Cumulative incidence functions were calculated at 100 days and 18 months based on time to IFD, using dates of IFD-free death, initiation of salvage treatment, and hematopoietic cell transplantation as competing risks. Eight patients (2.9%) developed IFD, including 3 Pneumocystis jirovecii pneumonia, 3 invasive mold infections (IMIs), and 2 invasive yeast infections (IYIs). The 100-day cumulative incidence of IFD accounting for competing risks was 1.8% (95% confidence interval [CI], 0.8% to 4.4%). Among the 280 patients, early toxicities including CRS (85%) and ICANS (55%) and late toxicities after day 30 including grades 3 and 4 neutropenia (41%) and low CD4 T-cell count (20%) were common. IFD was rare among patients who received CD19 CAR T-cell therapy for NHL in the absence of routine antifungal prophylaxis, despite frequent toxicities. These results suggest that, in settings with low institutional rates of IFD, routine antifungal prophylaxis may not be indicated.
Introduction
During recent years, CAR T-cell therapy has revolutionized the treatment of hematologic malignancies (HMs).1-6 Since 2017, four CD19 CAR T-cell products have been approved for the treatment of relapsed and refractory (R/R) non-Hodgkin lymphoma (NHL): axicabtagene ciloleucel (Yescarta; Gilead), tisagenlecleucel (Kymriah; Novartis), lisocabtagene maraleucel (Breyanzi; Juno), and brexucabtagene autoleucel (Tecartus; Kite).7,8 Ongoing clinical trials are evaluating the use of CAR T-cell therapy with novel targets for an array of other malignancies.9 Despite its efficacy in R/R HM, CAR T cells can cause significant toxicities.10 Cytokine release syndrome (CRS) and immune effector cell–associated neurotoxicity syndrome (ICANS) are two of the most serious direct toxicities that can manifest after cellular infusion, leading to critical illness and end-organ dysfunction.8,11,12 Although the administration of anti-inflammatory treatments, such as tocilizumab, an interleukin 6 (IL-6) receptor antagonist, and corticosteroids, have been shown to improve associated symptoms, these treatments contribute to additional immunosuppression during the first 30 days after cell infusion and have been identified as a risk factor for infectious complications in a patient population that is heavily immunosuppressed.10,13,14
Patients who receive CAR T-cell therapy have multiple defects in immunity that may predispose them to infection. When evaluating infections after CAR T-cell therapy, it is important to note that patients experience distinct periods of risk based on the associated immunologic defects. Preceding antitumor regimens, failed hematopoietic cell transplant (HCT), and advanced baseline disease affect patients’ early and late risk for infections after CAR T-cell therapy.15 Lymphodepleting chemotherapy with fludarabine and cyclophosphamide leads to an early period of high risk of infection with associated neutropenia. Additional therapies, including corticosteroids and tocilizumab for treatment of CRS and ICANS, may add to this early period of risk.15 A later risk period is driven by the off-tumor, on-target effects of the CAR T cells, with prolonged B-cell aplasia and associated hypogammaglobulinemia.15 Prolonged cytopenias up to 6 to 12 months after infusion have also been reported in 16% to 30% of patients who undergo CAR T-cell therapy.16-18 Further investigation of the mechanisms driving late neutropenia and T-cell depletion are needed.
Data regarding the infectious complications of CAR T-cell therapy are still somewhat limited and are influenced by institutional practices including variable use of antimicrobial prophylaxis.13,14,16,17,19 -21 Thus far, reports indicate that the incidence of infections is greatest in the first 30 days after CAR T-cell therapy, although late infections can be seen.13,14,19 Bacterial infections are the most common infectious complication in the first month, and viral infections are more frequently seen after 30 days.13,14 Invasive fungal disease (IFD) and in particular, invasive mold infections (IMIs) have been reported to be rare.13,14,16,19,20,22-24
Currently, optimal management of infectious prophylaxis including antiyeast and antimold prophylaxis is debated.15,20,22,25,26 Antifungal prophylaxis, when used in the right context, may prevent substantial morbidity related to IFD.27-32 However, these agents also cause toxicities, often have important drug interactions, and drive antifungal resistance.33-35 In this study, we sought to determine the incidence of IFD after CAR T-cell therapy in the absence of routine antifungal prophylaxis and to inform clinical guidelines for prophylaxis vs preemptive therapy.
Methods
Patients
We performed a single-center retrospective cohort study of sequential adults (age, ≥18) undergoing treatment of large B-cell lymphoma, follicular lymphoma, and mantle cell lymphoma with commercially available CD19 CAR T-cell therapy from December 2017 through September 2021 (N = 280) at Brigham and Women’s Hospital and Dana-Farber Cancer Institute (Boston, MA). The study was approved by the Dana-Farber Office of Human Research Studies and Institutional Review Board. Patients were identified through the Dana-Farber Cancer Institute Commercial Immune Effector Cells Database.
Cellular therapy clinical protocols
Patients received lymphodepleting chemotherapy with fludarabine 30 mg/m2 and cyclophosphamide 500 mg/m2 on days −3 to −5. Commercially available CD19-directed CAR T cells were administered at the US Food and Drug Administration (FDA)–approved dose, per institutional protocols. CRS and ICANS were treated according to the American Society of Clinical Oncology guidelines for immune-related adverse events after CAR T-cell therapy.36 Unless contraindicated, all patients received herpes virus prophylaxis with acyclovir and Pneumocystis jirovecii pneumonia (PJP) prophylaxis with trimethoprim-sulfamethoxazole or atovaquone from day 0 to at least day 180. Patients also received levofloxacin antibacterial prophylaxis from day 0 until count recovery, with absolute neutrophil counts (ANCs) ≥500. Routine antiyeast or antimold prophylaxis was not administered. Patients were treated using a preemptive febrile neutropenia protocol where empiric anti-Pseudomonas Gram-negative antibacterial therapy, including cefepime or ceftazidime, were initiated at first onset of febrile neutropenia. Empiric antifungal therapy with micafungin was initiated for persistent neutropenic fevers ≥4 days after the start of treatment with empiric antibacterials or for recurrent fevers after defervescence during neutropenia.
Definition of invasive fungal disease
IFD was identified between day of cell infusion (day 0) to last follow-up. Proven or probable IFD was defined according to the Revised European Organization for Research and Treatment of Cancer and the Mycoses Study Group (EORTC/MSG) criteria (September 2020).37,38 Only proven and probable infections were included. The date of diagnosis was recorded as the day of first positive microbiologic or histopathologic test results.
Late toxicities
Late hematologic toxicities and hypogammaglobulinemia were recorded for the cohort, with any ANC <1000, IgG <400, or CD4 T-cell count <200 recorded for 3 time periods: days 30 to 100, days 100 to 365, and after day 365 until last follow-up. Patients were also assessed for receipt of intravenous immunoglobulin at any time point after day 30.
Data collection and statistical analysis
Data collected from the electronic medical record included baseline characteristics such as age, sex, underlying disease, date of disease diagnosis, and history of prior autologous or allogeneic HCT. Data were also collected on exposure to potential mediators of IFD and CAR-T outcomes, including development of CRS, maximum grade CRS, administration of tocilizumab for CRS, number of doses of tocilizumab, administration of corticosteroids for CRS, diagnosis of ICANS, maximum grade ICANS, administration of corticosteroids for ICANS, post–CAR-T hypogammaglobulinemia, late neutropenia (>30 days after infusion), CD4 <200 (>30 days after infusion), disease progression, and death. CRS was defined according to the modified Lee criteria.11 ICANS was graded according to the Common Terminology Criteria for Adverse Events version 5 (CTCAE, v5z) criteria.11,39 Patients were censored on the first day of any additional antineoplastic therapy after disease relapse. Patients who did not experience relapse were observed until death or last follow-up until 25 January 2022.
Baseline characteristics and outcomes after CAR T-cell therapy were reported using summary statistics for categorical variables and medians and ranges for continuous variables. Overall cumulative incidence functions at 100 days and 18 months were calculated based on time to IFD, with dates of IFD-free death, initiation of salvage treatment after disease relapse, and repeat HCT used as competing risks. Analyses were performed with SAS Studio, version 9.04.01M6.
Results
Overall, 280 consecutive patients received commercial CD19 CAR T-cell therapy from December 2017 through September 2021. The median duration of follow-up was 259 days (range, 7-1340; interquartile range, 97-533). Baseline characteristics are described in Table 1. The median age was 64 (range, 19-82); 66% were male and 34% were female. All patients had non-Hodgkin lymphoma; the majority of cases (56%) were diffuse large B-cell lymphoma. Other common NHL diagnoses included in the cohort are shown in Table 1. Patients had received a median of 3 prior lines of chemotherapy (range, 2-10). Approximately one-third (33%) of patients had undergone prior HCT, most of which were autologous (90%). The majority of patients (87%) received axicabtagene ciloleucel (n = 244).
Early CAR T-cell toxicities are shown in Table 2. Cytokine release syndrome was observed in 239 patients (85%) after cell infusion. Maximum grades 1 (n = 102; 43%) and 2 (n = 125; 52%) CRS were most common, whereas maximum grades 3 (n = 8; 3%) and grade 4 (n = 4; 2%) were less frequently observed. Seventy-five percent of patients with CRS were treated with tocilizumab, with a median of 2 doses administered (range, 1-4). Corticosteroids were administered for treatment of CRS in approximately half of those who developed the syndrome (n = 118; 49%), with a median of 2 doses (range, 1-18) of 10 mg (mg) dexamethasone administered, and in 7 patients who received high-dose methylprednisolone. ICANS was observed in 153 patients (55%) after infusion of cells, with the median occurrence on day 6 (range, 0-34). When ICANS occurred, it was often severe, with grade 3 disease most frequently diagnosed (n = 86; 56%). Corticosteroids were administered for ICANS in 67% of those with a reported diagnosis, with a median of 5 doses (range, 1-90) of 10 mg dexamethasone and 10 patients who received high-dose methylprednisolone.
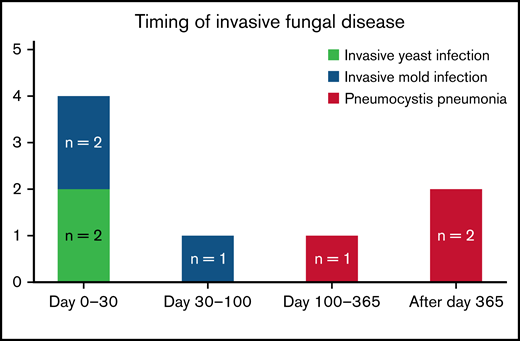
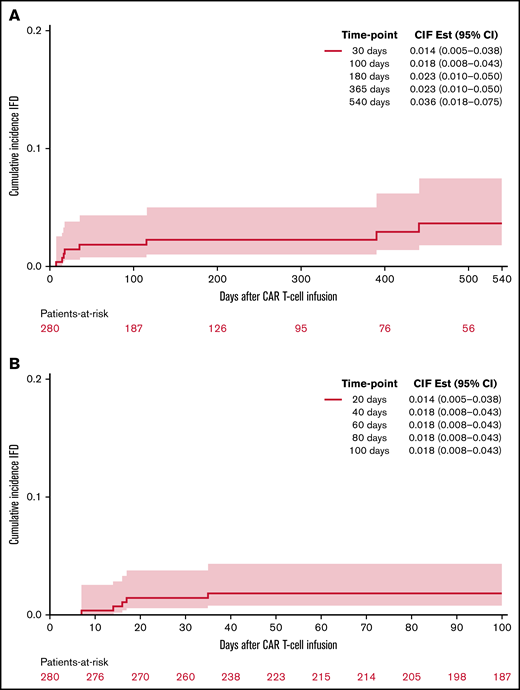
Invasive fungal disease was rare after CD19 CAR T-cell therapy despite the lack of routine antiyeast prophylaxis during the period of neutropenia. Eight patients (2.9%) overall developed IFD after cell infusion. Five patients developed early IFD before day 100 (3 IMIs; 2 IYIs) at a median time to onset of 16 days (range, 7-35), and 3 patients developed late IFD with PJP at a median time to onset of 390 days (range, 115-441). The characteristics and timing of the reported IFD are shown in Figure 1, and the individual cases are described in Table 3. Two patients (0.7%) developed IYIs, both of which were proven (Candida albicans empyema and C tropicalis fungemia) and occurred early after CAR T-cell therapy (days 16 and 17) in patients with central venous catheters. Both isolates were susceptible to fluconazole. Two patients developed probable invasive pulmonary aspergillosis: 1 had Aspergillus fumigatus isolated in a respiratory culture, 1 had a positive serum galactomannan >1.0, and both had corresponding radiographic findings and host risk factors. One patient developed a probable Rhizopus pulmonary infection, based on respiratory culture, radiographic findings, and host risk factors. These 3 IMIs (1.1%) also occurred early after CAR T-cell therapy (days 7, 13, and 35). The 100-day cumulative incidence of IFD accounting for competing risks was 1.8% (95% confidence interval [CI], 0.8% to 4.4%; Figure 2). Of note, 45 patients were lost to follow-up before day 100. Three additional patients (1.1%) were diagnosed with PJP after day 100 (days 115, 390, and 441). Two of the patients were not receiving PJP prophylaxis because of the timing (>1 year after CAR T-cell therapy). One patient had been prescribed atovaquone but had not been taking it leading up to diagnosis. The 18-month cumulative incidence of IFD accounting for competing risks was 3.8% (95% CI, 1.8-7.8) with 117 patients lost to follow-up by day 540 (Figure 2).
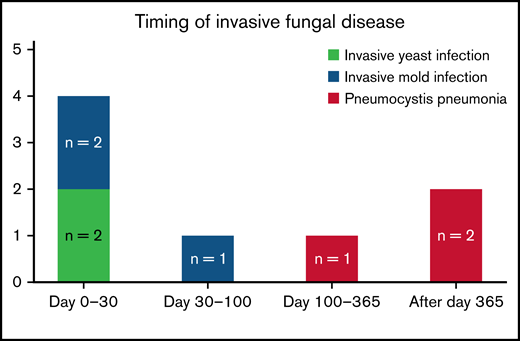
Timing and characteristics of invasive fungal disease after CAR T-cell therapy.
Timing and characteristics of invasive fungal disease after CAR T-cell therapy.
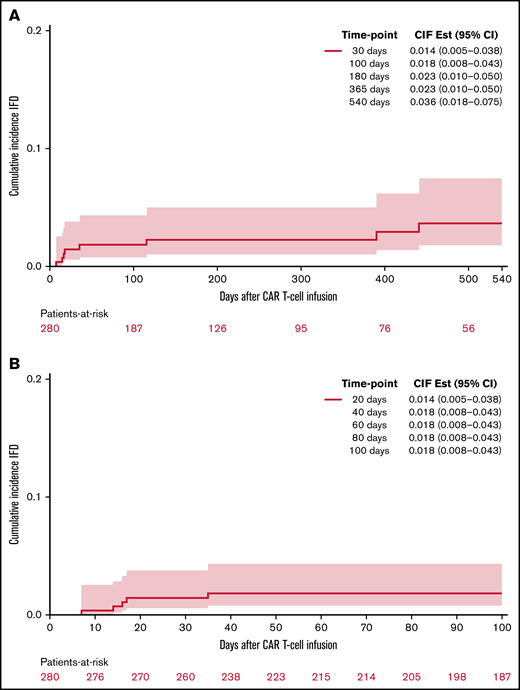
Cumulative incidence of proven or probable invasive fungal disease following CD19 CAR T-cell therapy. At 18 months (A) and 100 days (B).
Cumulative incidence of proven or probable invasive fungal disease following CD19 CAR T-cell therapy. At 18 months (A) and 100 days (B).
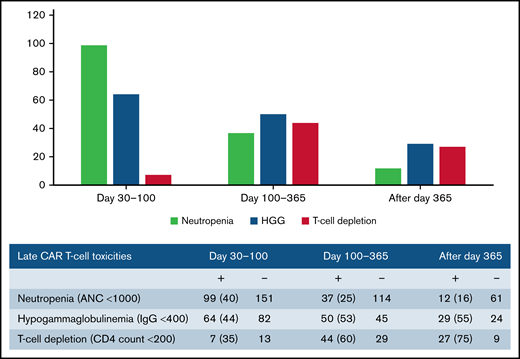
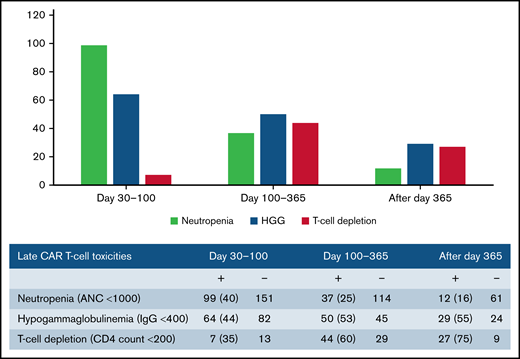
Late effects of CAR T-cell therapy include hematologic toxicity and hypogammaglobulinemia. We evaluated for these late toxicities at multiple stages: days 30 to 100, days 100 to 365, and after day 365. Table 4 demonstrates post-CAR T-cell outcomes. Overall, 116 patients (41%) experienced grade 3 and 4 neutropenia after day 30, 99 patients (35%) had documented hypogammaglobulinemia with IgG <400, and 55 patients (20%) had a CD4 count <200 after day 30 following CAR T-cell infusion. Neutropenia was more common from days 30 to 100, whereas hypogammaglobulinemia was more frequent after day 100 (Figure 3). In terms of other CAR T-cell outcomes, 122 patients (44%) experienced disease progression at a median of 83 days after cell infusion (range, 7-742). Sixty-eight patients (24%) died a median of 150 days after cell infusion (range, 7-664); the majority of the deaths (74%) were related to primary disease. One patient died of septic shock in the setting of C. tropicalis fungemia. No other patients had IFD as the primary cause of death.
Discussion
To our knowledge, this is the largest study to date in which the incidence and characteristics of IFD after CD19 CAR T-cell therapy were evaluated (N = 280) and is unique, in that the study population did not receive routine antiyeast or antimold prophylaxis. Despite high rates of CRS, ICANS, and late hematologic toxicities, including grade 3 and 4 neutropenia, hypogammaglobulinemia, and CD4 T-cell counts <200, a few cases of IFD were identified (n = 8), with an 18-month cumulative incidence of 3.8% (95% CI,1.8-7.8). The decision to use antimicrobial prophylaxis is nuanced and should be tailored to population risks, individual risks, and institutional incidence of the infectious outcome, as well as consideration of the harms and benefits of the prophylaxis agents.26,27,40 Given the potential disadvantages of antifungal prophylaxis, including toxicities, drug interactions, antifungal resistance, fungal dysbiosis, and breakthrough fungal infections, and the low overall incidence of IFD in this cohort, the results of this study suggest that routine antifungal prophylaxis may not be needed for patients undergoing treatment with CAR T-cell therapy for NHL in settings with a low institutional incidence of IFD.34-36,42-44 Although recent CAR-T-cell–specific guidelines recommend the routine use of antiyeast prophylaxis with fluconazole during the period of neutropenia after CAR T-cell therapy, it is noteworthy that only 1 yeast infection (0.4%) during the period of neutropenia potentially would have been prevented by fluconazole prophylaxis in a cohort of 280 patients.8,15,44 The use of antimold prophylaxis after CAR T-cell therapy is also a topic of ongoing debate.20,26 We also demonstrated a low rate of IMI (1.1%) after CD19 CAR T-cell therapy that would not justify the use of routine antimold prophylaxis in our patient population.20,27 Our data suggest that a measured approach to prophylaxis that reflects the institutional rates of IFD may be an acceptable alternative to a universal antifungal prophylaxis strategy. Further investigation into unique risk factors for IFD may also permit targeted strategies for prophylaxis in individual patients.
Among the 280 patients that were observed until last follow-up, death, or salvage antineoplastic therapy after disease relapse, only 8 developed proven or probable IFD. Although the small samples preclude risk factor analysis, important insights may be gained from the individual cases. Invasive yeast infections were rare in this cohort (n = 2), but occurred early after CAR T-cell infusion (days 16 and 17). Both patients with yeast infections and 3 of 4 (75%) of those with early IFD (days 0-30) developed CRS and ICANS requiring treatment with tocilizumab and corticosteroids before diagnosis of IFD. Multiple studies have shown an association between CRS and/or ICANS and risk of infection, although it remains unclear whether this finding is driven by the treatments including corticosteroids and tocilizumab or by the disease process itself.13,14,16,19 No studies have specifically evaluated the association between CRS and/or ICANS and invasive fungal disease; further investigation of this potential risk factor may help to guide decisions on prophylaxis or preemptive therapy.
Three patients in our cohort developed IMIs, most of which occurred early after CAR T-cell infusion (days 7, 13, and 35). One of those patients with probable Rhizopus pulmonary infection had aggressive diffuse large B-cell lymphoma that progressed to hemophagocytic lymphohistiocytosis with profound cytopenias and treatment with high-dose corticosteroids and etoposide in the weeks preceding CAR T-cell therapy. The other 2 patients with probable invasive pulmonary aspergillosis had an underlying diagnosis of transformed chronic lymphocytic lymphoma (tCLL) and represent 2 of only 7 (29%) patients with tCLL in the cohort. Notably, both patients had received ibrutinib in the weeks preceding CAR T-cell therapy, a potential independent risk factor for invasive mold infections.45-47 One of these patients also continued on ibrutinib after CAR T-cell therapy. Pre-CAR-T-cell patient characteristics most likely play a fundamental role in the risk for invasive mold infections after CAR T-cell therapy.15,24,48 Patients with CLL or tCLL may have unique risk factors for IFD related to baseline immune defects and prior CLL-specific therapies such as ibrutinib. In a recent review of IFD after CAR T-cell therapy, the majority of IMI cases (11/15; 73%) occurred in patients with B-cell acute lymphoblastic leukemia or CLL.24 Notably, our cohort does not include patients with B-cell acute lymphoblastic leukemia, which is a limitation and may partially explain the lower rate of IMI in this cohort. Further studies are needed to assess the IFD risk between unique disease groups and to evaluate the impact of pre-CAR-T-cell therapies on post-CAR-T-cell invasive fungal disease.
Prolonged neutropenia is a known risk factor for invasive fungal disease in patients with HM.29,49 , -53 Late neutropenia after day 30 has been reported after CAR T-cell therapy and can occur in a biphasic pattern.17,18,54 Although the mechanism is unknown, it has been theorized that it may be driven by perturbations in chemokines, and several studies have found an association with severe CRS.17,18,55 In their review, Haidar et al suggested a framework to assess risk of IFD after CAR T-cell therapy that delineates patients with prolonged neutropenia as having an “AML-like” phenotype and suggests that antimold prophylaxis may be considered in these patients.20 Herein, we report a high prevalence of late grade 3 and 4 neutropenia (41%) after day 30. Of those patients with ANC measured after day 30, 40% had neutropenia from days 30 to 100, 25% from days 100 to 365, and 16% after day 365. Despite these findings, there were no cases of IMI after day 35, indicating that late neutropenia alone without other risk factors may not lead to significant incidence of IFD in this population. Antimold prophylaxis after day 35 in patients with neutropenia in our cohort would have prevented no mold infections.
Three patients developed PJP late after CAR T-cell therapy (days 115, 390, and 441) after cessation of anti-Pneumocystis prophylaxis. One patient diagnosed at day 115 had been prescribed atovaquone prophylaxis at the time of discharge but did not fill the prescription. His labs were notable for lymphopenia when admitted with PJP. The second patient diagnosed at day 390 had hypogammaglobulinemia and primary adrenal insufficiency requiring total 20 mg hydrocortisone daily. Atovaquone prophylaxis was stopped at day 278, given clinical stability and a CD4 T-cell count >200. The third patient diagnosed at day 441 had hypogammaglobulinemia and a CD4 T-cell count <200 preceding the diagnosis. The patient had stopped taking TMP/SMX prophylaxis 2 months before presentation. In this cohort, CD4 T-cell counts were not consistently checked in all patients after CAR T-cell therapy, but were often <200 when checked. The clinical significance of this finding in the CAR T-cell patient population is unknown, and it remains unclear whether or how CAR T-cell therapy may elicit this immunologic effect or if this may be related to preceding antitumor therapies or HCT. Nonetheless, these late cases of PJP imply a prolonged immunosuppressed state in a subset of patients that merits further investigation and has been identified in other cohorts as well.14,19,49,56 Clinicians should consider whether extension of anti-Pneumocystis prophylaxis up to 1 year or use of CD4 count >200 as a requirement for cessation of prophylaxis may reduce late cases. Improved understanding of immune reconstitution after CAR T-cell therapy may aid in identifying which patients remain at risk for late infections and allow for tailored recommendations on duration of antimicrobial prophylaxis.
This study was limited by its retrospective nature, as well as the low number of cases of IFD that precluded risk factor analysis. Furthermore, this cohort included only patients with NHL, and it is possible that patients with ALL and CLL receiving CAR T-cell therapy would have a higher baseline rate of IFD related to their prior treatments or baseline state of immunosuppression. This consideration should be included when making decisions regarding antifungal prophylaxis. Although all patients were observed through their initial CAR T-cell–related hospitalization, which includes the highest period of risk for infections and the recommended period of antiyeast prophylaxis, many patients subsequently returned to care with local oncologists and were lost to follow-up including 45 patients before day 100 and 117 patients before day 540. Thus, late cases of IFD may not have been captured in those patients and should be considered when assessing the data. Finally, although our findings may not apply to centers with high institutional rates of IFD, they most likely will provide further insight for centers where the rates of IFD are low and may encourage institutions to reconsider the potential harms and benefits of routine antiyeast and antimold prophylaxis.
In our study, we investigated the incidence and characteristics of IFD in a large cohort (N = 280) of patients with NHL receiving CD19 CAR T-cell therapy. Despite a lack of routine antiyeast or antimold prophylaxis and frequent toxicities including CRS, ICANS, and late neutropenia, the incidence of IFD was low. Furthermore, several of these infections occurred very early after cell infusion, emphasizing the fundamental importance of a patient’s baseline risk factors in determining their post–CAR-T-cell IFD risk. With the rising threat of widespread antifungal resistance, stewardship around prophylaxis and continued efforts to target these therapies to the patients at the highest risk is vital.57,58 Future important avenues for investigation include improved characterization of immune reconstitution after CAR T-cell therapy and identification of key risk factors for IFD that may permit individualized strategies for antimicrobial prophylaxis and treatment.
Acknowledgment
The authors acknowledge the inspiration and guidance that Francisco Marty provided for this project.
Francisco M. Marty died on 8 April 2021.
Authorship
Contribution: J.S.L. designed the research, collected and analyzed the data, and wrote the manuscript; M.M.A. and I.H.G.-B. collected the data and edited the manuscript; K.B. assisted with regulatory processes and edited the manuscript; F.M.M. designed the research; J.M.G. and S.K. assisted in designing the study and edited the manuscript; S.P.H. designed the research and edited the manuscript; C.A.J. assisted in data collection, designed the study, and edited the manuscript.
Conflict-of-interest disclosure: C.A.J. has been a consultant to Kite/Gilead, Novartis, BMS/Celgene, Bluebird Bio, Epizyme, Ipsen, Lonza, Instill Bio, and Abintus Bio and has received research funding from Kite/Gilead and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Jessica S. Little, Brigham and Women’s Hospital, 75 Francis St, PBB-A4, Boston, MA 02115, e-mail: jlittle@bwh.harvard.edu.