Key Points
First description of a patient with a germline GATA2 mutation and diagnosis of primary myelofibrosis.
Development of bone marrow failure on a Janus kinase inhibitor.
Introduction
The human GATA2 gene is located on the long arm of chromosome 3 (3q21.3) and contains 6 to 7 exons.1 Together with GATA1 and GATA3, it is primarily expressed in hematopoietic cells and is a master regulator of early hematopoiesis.2-8
Mutations in GATA2 can be either inherited or acquired. The GATA2-deficiency syndrome was first described as an autosomal-dominant immunodeficiency and bone marrow (BM) failure disorder caused by heterozygous loss-of-function mutations in GATA2.9-15 It is also an established cause of familial forms of acute myeloid leukemia (AML) and myelodysplastic syndrome.14 Furthermore, it is associated with an increased risk of chronic myelomonocytic leukemia.16 In patients with GATA2 deficiency, the BM examination typically reveals hypocellularity with a severe decrease of hematogones, monocytes, natural killer cells, and B cells. Atypical megakaryocytes are frequently found.10,17 Recently, single-cell analysis of GATA2-deficient patients revealed a markedly skewed differentiation pattern with expansion of the erythroid and megakaryocytic lineage and a reduction in myeloid and lymphoid differentiation.18
Acquired GATA2 mutations are found in patients with myeloid neoplasms. In AML, somatic GATA2 gain-of-function mutations are associated with mutations in FLT3 and NPM1 and correlate with poor outcome.19,20 In chronic myeloid leukemia, a gain-of-function mutation in GATA2 (p.Leu359Val) was found in 10% of cases during blast phase.21 In contrast, <5% of patients with primary myelofibrosis (PMF) carry somatic GATA2 mutations.22 Although GATA1 downregulation in megakaryocytes was observed in PMF, there is no established association between GATA2-deficiency syndrome and PMF.23 Here, we describe, to our knowledge, the first case of a patient with PMF and an inherited GATA2 mutation who developed BM failure on a Janus kinase (JAK) inhibitor.
Case description
Before the diagnosis of PMF at the age of 38 years, the female patient had suffered an abortion and had a history of human papillomavirus–positive cervical dysplasia. At diagnosis of PMF, the patient presented with asymptomatic hepatosplenomegaly. Her complete blood count revealed mild thrombocytopenia with normal hemoglobin and leukocyte values (Figure 1). The peripheral blood smear showed abundant erythrocyte teardrops and leukoerythroblastosis. The lactate dehydrogenase was markedly elevated. The BM biopsy was hypercellular, with increased inconspicuous granulopoiesis, preserved erythropoiesis, and marked proliferation of pleomorphic megakaryocytes, with a predominance of large forms with hyperchromatic, bulbous, and naked nuclei (Figure 2A). There were occasional small megakaryocytes with hyposegmented nuclei as well as some large forms with multiple separated nuclei. Most of the megakaryocytes formed clusters and sheets, frequently adjacent to bone trabeculae within dilated vascular sinuses. A BM fibrosis grade 2 was diagnosed according to World Health Organization (WHO) criteria and a JAK2-V617F mutation was detected by qualitative polymerase chain reaction. In addition, conventional cytogenetics identified a translocation of chromosomes 3 and 12 (46,XX,t(3;12)(p21;p13)[3]/46,XX[7]).
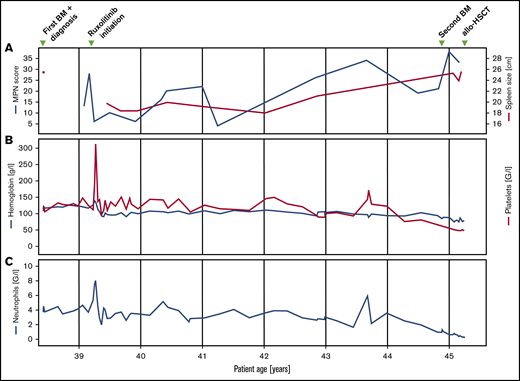
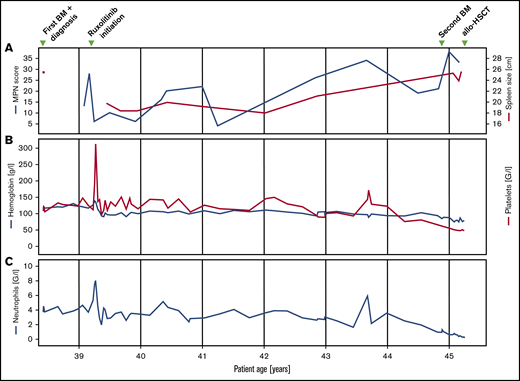
Clinical and laboratory parameters. Longitudinal follow-up of myeloproliferative neoplasm total symptom score, spleen size (A), hemoglobin, platelet (B), and neutrophil count (C).
Clinical and laboratory parameters. Longitudinal follow-up of myeloproliferative neoplasm total symptom score, spleen size (A), hemoglobin, platelet (B), and neutrophil count (C).
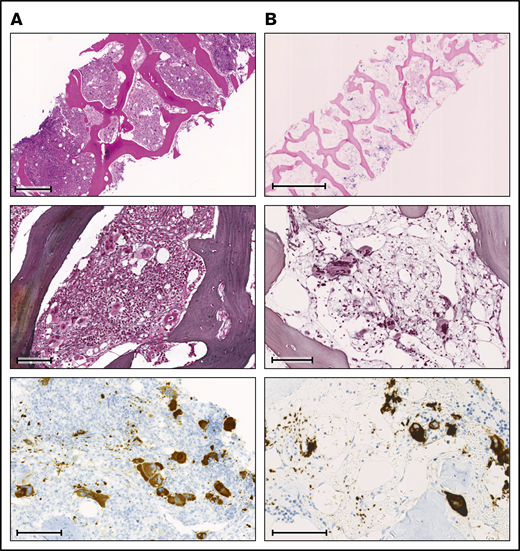
Comparison of BM biopsies at diagnosis and during follow-up. (A) BM biopsy at diagnosis. (B) BM biopsy at follow-up 6 years later. Row 1: (A) H&E stain; scale bar, 500 μm. (B) Giemsa stain; scale bar, 1000 μm. Row 2: (A-B) Gomori methenamine silver stain; scale bar, 100 μm. Row 3: (A-B) CD61 immunohistochemistry; scale bars, 100 μm.
Comparison of BM biopsies at diagnosis and during follow-up. (A) BM biopsy at diagnosis. (B) BM biopsy at follow-up 6 years later. Row 1: (A) H&E stain; scale bar, 500 μm. (B) Giemsa stain; scale bar, 1000 μm. Row 2: (A-B) Gomori methenamine silver stain; scale bar, 100 μm. Row 3: (A-B) CD61 immunohistochemistry; scale bars, 100 μm.
Fluorescence in situ hybridization analysis confirmed the loss of one 3′ETV6 signal (centromeric probe) in 50 of 200 nuclei (25%). Based on the WHO 2016 classification, the diagnosis of PMF was made. Three months after the initial diagnosis, the patient reported satiety and symptomatic splenomegaly. The spleen was palpable 10 cm below the left costal arch and the myeloproliferative neoplasm (MPN) total symptom score24 was 15. Over the next 8 months, fatigue and symptomatic splenomegaly worsened and the patients also reported night sweats. A therapy with the JAK inhibitor ruxolitinib was initiated and her symptoms responded promptly (Figure 1). Six months after therapy initiation, the spleen size was reduced to 3 to 4 cm below the left costal arch. On therapy, the patient suffered recurrent cutaneous herpes zoster infections. Three years after treatment initiation, PMF-associated symptoms deteriorated and the spleen size increased, implying resistance to ruxolitinib. Due to the lack of alternative treatment options including allogeneic hematopoietic stem cell transplantation (allo-HSCT), which the patient declined, she remained on a stable dose of ruxolitinib. Six years after the initial diagnosis, the patient was diagnosed with basal cell carcinoma of the facial skin. Coincidentally, pancytopenia with anemia grade 2, thrombocytopenia grade 2, and new-onset neutropenia grade 3 were diagnosed. The dose of ruxolitinib was reduced, which led to further worsening of the symptoms, but no improvement of the blood counts, which continuously decreased (minimal values: hemoglobin, 68 g/L; platelets, 38 × 109/L; neutrophils 0.23 × 109/L). A follow-up BM examination was performed (Figure 2B). In contrast to the BM analysis, at diagnosis 6 years prior, the cellularity was markedly reduced, particularly myelopoiesis. Megakaryopoiesis remained increased with typical PMF morphology. As at diagnosis, some small and hyposegmented/monosegmented and larger multinucleated forms were noted. Next-generation sequencing analysis revealed the known JAK2-V617F mutation (variant allele frequency [VAF], 80%) and a GATA2-N317S mutation (VAF, 48%). The molecular analysis of the buccal swab and of the biopsy of the cervix uteri confirmed the germline origin of the GATA2 (VAF, 51.6% [buccal swab] and 50.0% [cervix uteri]), but not the JAK2, mutation (VAF, 5.5% [buccal swab] and 11.8% [cervix uteri]). The diagnosis of BM failure in the context of GATA2 deficiency was made and the patient proceeded to allo-HSCT.
At the last follow-up, 9 months after allo-HSCT, the patient was in a complete molecular remission with a complete donor chimerism (>99%). The BM fibrosis was strongly reduced with only a minimal increase of reticulin fibers.
Methods
Patient and healthy donor samples
Peripheral blood samples were collected after obtaining informed consent.
Next-generation sequencing analysis
Genomic profiling was performed by using the Illumina TruSight Myeloid, Archer VariantPlex Myeloid and FoundationOne Heme Panels.
Flow cytometry
Mobilized peripheral blood mononuclear cells (PBMCs) from a healthy donor (control) and PBMCs from a patient with PMF were stained for NK cells with the following monoclonal antibodies: Zombie Aqua Fixable Viability Kit (BioLegend), PC5.5-Conjugated Anti-Human CD56 (clone N901; Beckman Coulter), and Pacific Blue-Conjugated Anti-Human CD3 (clone UCHT1; Beckman Coulter). Stained cells were analyzed on a BD LSRFortessa cell analyzer.
Results and discussion
We retrospectively performed a targeted sequencing analysis of genes frequently mutated in myeloid neoplasms at diagnosis and at the BM failure stage 6 years later. The burden of the mutated JAK2 allele rose from 55% at diagnosis to 80%, whereas, as predicted, the VAF of the GATA2 mutation remained stable (supplemental Table 1). The increase of the mutated JAK2 allelic burden was paralleled by an increase of the t(3;12)(p21;p13) mutated clone, which leads to rearrangement of ETV6. ETV6 rearrangements have been described in 0.3% of patients with MPNs, but not in PMF.25-27 The germline GATA2-N317S mutation is a missense mutation that has previously been described in patients with myelodysplastic syndrome and AML but not in patients with GATA2-deficiency syndrome. Although the familial history did not reveal stigmata of GATA2 deficiency, this patient had suffered several manifestations of GATA2-deficiency syndrome; spontaneous abortion, recurrent human papillomavirus, and herpes virus infections have been reported. Also, patients with germline GATA2 mutation are at increased risk for skin cancer, including basal cell carcinoma. Moreover, we detected intermittent monocytopenia, but no deficiency in natural killer cells (supplemental Figures 1-2).
First-degree relatives of patients with MPNs have a fivefold to sevenfold increased risk of MPN development.28 Recently, 17 genetic risk loci for MPN development were identified, including the previously reported JAK2 46/1 haplotype.29,30 Interestingly, 1 of these risk loci was detected in chromosomal locus 3q21.3 and the main single-nucleotide polymorphism (rs9864772) localized to a distal enhancer for GATA2. In the presented case, the GATA2 mutation preceded and possibly favored the acquisition of a JAK2 mutation, which led to PMF development. Previous studies have shown that mutations can precede the acquisition of a JAK2 mutation in MPNs.31
Cytopenias are common adverse events of ruxolitinib therapy, particularly at the beginning of treatment. However, new-onset grade 3/4 cytopenias after >3.5 years of treatment are unlikely to be ruxolitinib-related.32 They may suggest transformation to AML, which was excluded in this patient. The sudden development of late pancytopenia in this patient is therefore likely related to the GATA2-deficiency syndrome rather than to ruxolitinib treatment. We therefore propose to assess germline origin in patients with PMF and GATA2 mutations, as development of pancytopenia in PMF patients with germline GATA2 mutations may be indicative of BM failure. Of note, a patient with PMF who developed aplastic BM while treated with the JAK inhibitor fedratinib has previously been described, but molecular analyses were not reported.33
In summary, we here report the first case of a patient with GATA2-deficiency syndrome who developed PMF. The identification of germline GATA2 mutations in PMF is of particular relevance because patients may proceed to related donor allo-HSCT.
Requests for data may be e-mailed to the corresponding author, Alexandre P. A. Theocharides, at alexandre.theocharides@usz.ch.
Acknowledgment
A.P.A.T. was supported by the Professor Dr Max Cloëtta Foundation.
Authorship
Contribution: C.V.R., E.H., V.L., S.B., and A.P.A.T. analyzed the data; and C.V.R. and A.P.A.T. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Alexandre P. A. Theocharides, Department of Medical Oncology and Hematology, University Hospital Zurich–University of Zurich, Rämistr 100, 8091 Zurich, Switzerland; e-mail: alexandre.theocharides@usz.ch.