Key Points
A total of 137 patients with JMML were analyzed using DREAM.
We developed a robust DNA methylation test that is highly consistent with the array-based international consensus definition for JMML.

Abstract
Juvenile myelomonocytic leukemia (JMML) is a rare myelodysplastic/myeloproliferative neoplasm that develops during infancy and early childhood. The array-based international consensus definition of DNA methylation has recently classified patients with JMML into the following 3 groups: high (HM), intermediate (IM), and low methylation (LM). To develop a simple and robust methylation clinical test, 137 patients with JMML were analyzed using the Digital Restriction Enzyme Analysis of Methylation (DREAM), which is a next-generation sequencing–based methylation analysis. Unsupervised consensus clustering of the discovery cohort (n = 99) using DREAM data identified HM (HM_DREAM; n = 35) and LM subgroups (LM_DREAM; n = 64). Of the 98 cases that could be compared with the international consensus classification, 90 HM (n = 30) and LM (n = 60) cases had 100% concordance with DREAM clustering results. Of the remaining 8 cases comprising the IM group, 4 were classified as belonging to the HM_DREAM group and 4 to the LM_DREAM group. A machine-learning classifier was successfully constructed using a support vector machine (SVM), which divided the validation cohort (n = 38) into HM (HM_SVM, n = 18) and LM (LM_SVM; n = 20) groups. Patients with the HM_SVM profile had a significantly poorer 5-year overall survival rate than those with the LM_SVM profile. In conclusion, we developed a robust methylation test using DREAM for patients with JMML. This simple and straightforward test can be easily incorporated into diagnosis to generate a methylation classification for patients so they can receive risk-adapted treatment in the context of future clinical trials.
Introduction
Disruption of the epigenome is a common finding in cancer cells, often resulting in altered DNA methylation patterns that can be accurately assessed using DNA methylation analysis.1 Some aberrant DNA methylation patterns have been associated with specific genetic mutations that promote neoplasia, and these patterns can be used as biomarkers for disease progression.2
Juvenile myelomonocytic leukemia (JMML) is a rare myelodysplastic/myeloproliferative neoplasm that develops during infancy and early childhood.3 JMML is characterized by excessive myelomonocytic cell proliferation and hypersensitivity to granulocyte-macrophage colony-stimulating factor. We and other groups conducted a genome-wide methylation profiling of promoter-associated CpG sites using the Infinium Human Methylation 450K BeadChip (450K; Illumina, San Diego, CA), which enabled the identification of patients with JMML with a high-methylation (HM) profile.4–6 The HM profile correlated significantly with genetic markers predicting poor outcome, including PTPN11/NF1 gene mutations, ≥2 genetic mutations, an acute myeloid leukemia (AML)–type gene expression profile, and LIN28B overexpression.4 Furthermore, an array-based international consensus definition of DNA methylation was recently reported, categorizing patients with JMML into 3 groups: HM, intermediate methylation (IM), and low methylation (LM), in an international collaborative study.7 To incorporate methylation profiling into the clinical decision-making process and future clinical trials, a robust but simple and less labor-intensive method for evaluating methylation patterns in patients with JMML is needed.
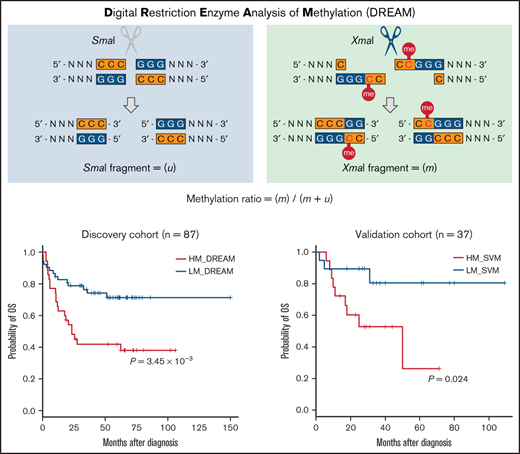
The Digital Restriction Enzyme Analysis of Methylation (DREAM) is a method for quantitative mapping of DNA methylation at 10s of thousands of CpG sites on a genome using a next-generation sequencing technology. Methylation levels at each of the target CpG sites are calculated by high-throughput sequencing of DNA fragments, with specific signatures for unmethylated and methylated CpG sites obtained by sequential digestion of genomic DNA using restriction enzymes SmaI and XmaI, which have the same CCCGGG recognition site but different sensitivity to CpG methylation and cleavage patterns.8
We report our progress toward establishing and implementing a method to evaluate DNA methylation using DREAM.
Patients and methods
Patients
In this study, DREAM was performed in 137 patients (JMML, n = 124; Noonan syndrome–associated myeloproliferative disorder [NS/MPD], n = 13) using genomic DNA from peripheral blood or bone marrow mononuclear cells. Patients were divided into 2 groups, a discovery cohort (n = 99) and a validation cohort (n = 38), and a diagnosis of JMML or NS/MPD was made based on internationally accepted criteria.9,10 Patients in the validation cohort were selected from 2 groups (Japan, n = 18; United States, n = 20) and had no previous DNA methylation classification or preselection according to known risk factors or parameters.
Written informed consent was obtained from the parents of all patients, and the study was approved by the ethics committee of Nagoya University Graduate School of Medicine.
Sample preparation
Genomic DNA was extracted from mononuclear cells derived from the peripheral blood or bone marrow of patients using the QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. All DNA samples were measured using Nanodrop 1000 (Thermo Fisher Scientific, Waltham, MA) to ensure that the A260/A280 ratios ranged between 1.8 and 2.0. Additionally, the DNA integrity number was also measured using the Agilent 2200 Tape Station System and the Agilent Genomic DNA Screen Tape Assay to ensure that DNA integrity number scores were >6.0 (Agilent, Santa Clara, CA).
Mutational analysis
Mutational analysis was performed in 99 patients from the discovery cohort using whole-exome sequencing (n = 60), targeted capture sequencing covering 184 genes (n = 2),11 and polymerase chain reaction (PCR) amplicon sequencing covering 8 genes (PTPN11, NRAS, KRAS, CBL, NF1, SETBP1, JAK3, and SH3BP1; n = 61). The same patients were also included in our previous cohort with unique patient numbers (UPNs) 1 to 152,4 and detailed descriptions of the methodology used and relevant genetic information have been provided elsewhere.4,12 In the validation cohort of 38 patients, canonical RAS pathway gene mutations (PTPN11, NRAS, KRAS, CBL, and NF1) were confirmed using Sanger sequencing (Japanese cohort, n = 18) or custom amplicon-based targeted sequencing (US cohort, n = 20), as previously described.13
The number of genetic mutations carried by each case was calculated by adding the number of driver gene mutations within (PTPN11, NF1, NRAS, KRAS, and CBL) and outside (ASXL1, IKZF1, JAK3, RRAS2, SETBP1, SH3BP1, and SOS1 and ALK/ROS1 fusions) the RAS pathway. Of the 99 patients, 16 (16.2%) harbored ≥2 driver gene mutations within and outside the RAS pathway, when chromosomal abnormalities were not counted in the number of gene mutations.
450K array analysis
Global DNA methylation profiles were analyzed in all patients in the discovery cohort using 450K, as previously reported and according to the manufacturer’s instructions.4
DREAM
DREAM was performed, as previously described.8 Briefly, 100 ng of genomic DNA extracted from JMML samples was sequentially cut using 2 restriction enzymes (SmaI and XmaI) recognizing the same sequence, 5'-CCCGGG-3' sites in DNA. Initially, genomic DNA was digested with 25 units of SmaI (New England Biolabs, Ipswich, MA) for 3 hours at 25°C. Thereafter, 12.5 units of XmaI (New England Biolabs) was added, and digestion was continued for an additional 16 hours at 37°C. The genomic DNA was then treated at 65°C for 1 hour to inactivate the restriction enzymes. SmaI does not cut methylated sites and leaves blunt ends, whereas XmaI can cleave methylated sites, leaving a 5' overhang sequence (Figure 1A), thus creating specific signatures for the methylated and unmethylated sites. The enzyme-treated DNA was then used to generate sequencing libraries according to protocols using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs), which repaired the ends of the DNA fragments and filled the 5' overhang sequences derived from XmaI digestion with complementary nucleotides.
![Performance evaluation of DREAM. (A) Schematic outline of the principles of DNA methylation analysis. In DREAM, DNA is sequentially cut using 2 restriction enzymes, SmaI and XmaI. Estimated durations are indicated alongside each step. Individual sample sequencing could be completed in 4 to 5 days, including SmaI digestion (3 hours), XmaI digestion (16 hours), end repair (2 hours), adaptor ligation (1 hour), PCR amplification (2 hours), final library quality control (1 day), sequencing (2-3 days), and data analysis time. The hands-on time was <6 hours per sample for the entire process. In reduced representation bisulfite sequencing (RRBS), DNA is digested by MspI to generate short fragments that contain CpG dinucleotides at the 3' ends. (B) In DREAM, the ends of the sequencing reads generated by SmaI or XmaI locate at 5'-CCCGGG-3' sites, with 4 types of read ends. Left-1, left-3, right-2, and right-4 can be caused by these restriction enzymes, whereas others can be caused by random fragmentation. Unmethylated 5'-CCCGGG-3' sites (presumed to be digested by SmaI) generate left-3 and right-2 ends (unmethylated signature [u]), and methylated 5'-CCC(me)GGG-3' sites (presumed to be digested by XmaI) generate left-1 and right-4 (methylated signature [m]). The methylation ratio was simply calculated as the fraction of total 5'-CCCGGG-3' site sequencing reads (m + u) that were mapped to m. (C) The human genome has 28.0 million CpG sites, and the GRCh37/hg19 annotation provides coordinates for 374 921 5'-CCCGGG-3' sites targeted by DREAM, including the 6455 sites covered by 450K. (D) Results of DREAM of 4 pg of methylation calibrators spiked in 100 ng of sample DNA from 19 patients are shown. EC247, EC293, and EC466 were used as unmethylated or 100% methylated calibration standards. (E) The median number of CpG sites and the median Pearson’s correlation coefficient (r2) between DREAM methylation ratios and 450K β values at 6455 overlapping CpG sites classified according to DREAM sequence depth for 19 cases are shown. Error bars represent standard errors. (F) Correlation of the discovery cohort (n = 99) between ratios obtained using DREAM and 450K β values at 1703 overlapping CpG sites with sufficient coverage (≥20 reads in 95% of 99 samples). The best (G) and worst (H) correlations are shown. TSS, transcription start site.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/5/24/10.1182_bloodadvances.2021005080/4/m_advancesadv2021005080f1.png?Expires=1769460040&Signature=twKPX7HqReu1pl-X8oFx-WMx~0mPfvxJ5453yqTx5RfkP3jksFy6ZDz6-Dqd2Ytwpnpmtcrtf1Z~~PokkQT47Lb395qvkKRc~OntYEAwL5F9IUhOb~vCDeZ-vQxm7JuBMC3WJgXOAsZrjC3TOeUooYfGC0a5fl2OPQrTA42zb~e4Qtu7LB0wyJ1uHayHi1~J-Q1Xy4hZBUdIAcfmcXOoFuYF8kA5XPGHJu0qu7cCtFprU9Jfz8tUWqSARSdqo-4IUKQv07dWU0l50-KBdugTa0mUQ2AJX3hz413lEL8FzYrcV1He1oAAnvnCFEwSe2srpbRjjfjBPx9daD8p5RRzDg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Performance evaluation of DREAM. (A) Schematic outline of the principles of DNA methylation analysis. In DREAM, DNA is sequentially cut using 2 restriction enzymes, SmaI and XmaI. Estimated durations are indicated alongside each step. Individual sample sequencing could be completed in 4 to 5 days, including SmaI digestion (3 hours), XmaI digestion (16 hours), end repair (2 hours), adaptor ligation (1 hour), PCR amplification (2 hours), final library quality control (1 day), sequencing (2-3 days), and data analysis time. The hands-on time was <6 hours per sample for the entire process. In reduced representation bisulfite sequencing (RRBS), DNA is digested by MspI to generate short fragments that contain CpG dinucleotides at the 3' ends. (B) In DREAM, the ends of the sequencing reads generated by SmaI or XmaI locate at 5'-CCCGGG-3' sites, with 4 types of read ends. Left-1, left-3, right-2, and right-4 can be caused by these restriction enzymes, whereas others can be caused by random fragmentation. Unmethylated 5'-CCCGGG-3' sites (presumed to be digested by SmaI) generate left-3 and right-2 ends (unmethylated signature [u]), and methylated 5'-CCC(me)GGG-3' sites (presumed to be digested by XmaI) generate left-1 and right-4 (methylated signature [m]). The methylation ratio was simply calculated as the fraction of total 5'-CCCGGG-3' site sequencing reads (m + u) that were mapped to m. (C) The human genome has 28.0 million CpG sites, and the GRCh37/hg19 annotation provides coordinates for 374 921 5'-CCCGGG-3' sites targeted by DREAM, including the 6455 sites covered by 450K. (D) Results of DREAM of 4 pg of methylation calibrators spiked in 100 ng of sample DNA from 19 patients are shown. EC247, EC293, and EC466 were used as unmethylated or 100% methylated calibration standards. (E) The median number of CpG sites and the median Pearson’s correlation coefficient (r2) between DREAM methylation ratios and 450K β values at 6455 overlapping CpG sites classified according to DREAM sequence depth for 19 cases are shown. Error bars represent standard errors. (F) Correlation of the discovery cohort (n = 99) between ratios obtained using DREAM and 450K β values at 1703 overlapping CpG sites with sufficient coverage (≥20 reads in 95% of 99 samples). The best (G) and worst (H) correlations are shown. TSS, transcription start site.
Performance evaluation of DREAM. (A) Schematic outline of the principles of DNA methylation analysis. In DREAM, DNA is sequentially cut using 2 restriction enzymes, SmaI and XmaI. Estimated durations are indicated alongside each step. Individual sample sequencing could be completed in 4 to 5 days, including SmaI digestion (3 hours), XmaI digestion (16 hours), end repair (2 hours), adaptor ligation (1 hour), PCR amplification (2 hours), final library quality control (1 day), sequencing (2-3 days), and data analysis time. The hands-on time was <6 hours per sample for the entire process. In reduced representation bisulfite sequencing (RRBS), DNA is digested by MspI to generate short fragments that contain CpG dinucleotides at the 3' ends. (B) In DREAM, the ends of the sequencing reads generated by SmaI or XmaI locate at 5'-CCCGGG-3' sites, with 4 types of read ends. Left-1, left-3, right-2, and right-4 can be caused by these restriction enzymes, whereas others can be caused by random fragmentation. Unmethylated 5'-CCCGGG-3' sites (presumed to be digested by SmaI) generate left-3 and right-2 ends (unmethylated signature [u]), and methylated 5'-CCC(me)GGG-3' sites (presumed to be digested by XmaI) generate left-1 and right-4 (methylated signature [m]). The methylation ratio was simply calculated as the fraction of total 5'-CCCGGG-3' site sequencing reads (m + u) that were mapped to m. (C) The human genome has 28.0 million CpG sites, and the GRCh37/hg19 annotation provides coordinates for 374 921 5'-CCCGGG-3' sites targeted by DREAM, including the 6455 sites covered by 450K. (D) Results of DREAM of 4 pg of methylation calibrators spiked in 100 ng of sample DNA from 19 patients are shown. EC247, EC293, and EC466 were used as unmethylated or 100% methylated calibration standards. (E) The median number of CpG sites and the median Pearson’s correlation coefficient (r2) between DREAM methylation ratios and 450K β values at 6455 overlapping CpG sites classified according to DREAM sequence depth for 19 cases are shown. Error bars represent standard errors. (F) Correlation of the discovery cohort (n = 99) between ratios obtained using DREAM and 450K β values at 1703 overlapping CpG sites with sufficient coverage (≥20 reads in 95% of 99 samples). The best (G) and worst (H) correlations are shown. TSS, transcription start site.
The prepared libraries were run on a HiSeq 2500 with 2 × 150 bp end reads (Illumina), and sequencing data were mapped to the hg19 human reference genome using the Burrows-Wheeler Aligner (https://github.com/lh3/bwa) with default and mem options. The end of the sequencing reads generated by SmaI or XmaI would locate at 5'-CCCGGG-3' sites, with 4 types of read ends, as shown in Figure 1B. The methylation ratio was simply calculated as the fraction of total 5'-CCCGGG-3' site sequencing reads (m + u) that were mapped to the methylated signature (m), using the bioinformatic scripts (supplemental script files [dreamtest.pdf and probefindtest.pdf]).
Calibration standards for DREAM
Calibration standards with defined methylation levels for 19 patients in the discovery cohort were added to the DREAM library. These standards consisted of PCR products created using unsheared Escherichia coli DNA as a template, as previously described.8 The sequences of the calibrators and primers used for making the PCR products (EC293, EC466, and EC247) are shown in supplemental Table 2. The unmethylated calibration standard consisted of the PCR products without further treatment, whereas the fully methylated calibration standard consisted of PCR products methylated using M.SssI CpG methyltransferase. The unmethylated and methylated standards of different sequences were mixed in a 1:1 ratio and were used as a methylation calibrator in subsequent experiments.
Clustering analysis
To perform clustering analysis, DREAM data were filtered, and sites with ≥20 reads in 95% of the 99 samples from the discovery cohort were included, resulting in 30 408 CpG sites. Of these, 7360 promoter-associated CpG sites located within 1000 bases from transcription start sites were used for bioinformatic analysis (supplemental Table 3).
For clustering analysis, missing values in each sample were imputed as the median of all nonmissing values for the corresponding CpG site. Unsupervised hierarchical clustering was performed according to the Canberra distance and Ward’s method (Ward.D2), as implemented by the hclust function in R (R Foundation for Statistical Computing, Vienna, Austria). The approved unbiased P value was calculated for each cluster using the bootstrap method of the R package, pvclust.15
SVM classifier construction
To construct an optimal support vector machine (SVM) classifier, the tune.svm function of the e1071 package in R was used.16
RRBS
RRBS was used to assess 10 patients with JMML.17 Briefly, purified genomic DNA was digested by the methylation-insensitive restriction enzyme MspI to generate short fragments that contained CpG dinucleotides at the ends. The CpG-rich DNA fragments (40-220 bp) were size selected, subjected to bisulfite conversion, PCR amplified, and end sequenced on an Illumina HiSeq2500 (Figure 1A). RRBS data were filtered to include sites with at least 10 reads in all samples, resulting in 4971 promoter-associated CpG sites located within 1000 bases from transcription start sites for bioinformatic analysis.18 Unsupervised hierarchical clustering was performed according to the Canberra distance and Ward.D2, as implemented by the hclust function in R.
RNA sequencing and expression analysis
Gene ontology and pathway analysis
Gene ontology annotations and the Kyoto Encyclopedia of Genes and Genomes pathway of the differentially expressed genes were performed using the DAVID (Database for Annotation, Visualization, and Integrated Discovery) online tool.22
Statistical analysis
Pearson’s (r2) and Spearman’s (ρ) correlation coefficients were used to measure the degree of association between 2 variables, including methylation ratios and β values. For unsupervised hierarchical clustering, the Canberra distance and Ward.D2 were used, as implemented by the hierarchical clustering (hclust) function in R. For comparison of the frequency of mutations and other clinical features between groups, categorical and continuous variables were analyzed using Fisher’s exact and Mann-Whitney U tests, respectively. Overall survival (OS), defined as time from date of diagnosis to death, and transplantation-free survival (TFS), defined as time from date of diagnosis to transplantation or death resulting from any cause, were estimated using the Kaplan-Meier method. The difference in survival was tested using the log-rank test, and hazard ratios for survival with 95% confidence intervals (CIs) were estimated according to the Cox proportional hazards model. To identify predictors of survival, we performed multivariable and univariable Cox regression analyses. Stepwise variable selection based on Akaike’s information criterion was used for the selection of variables in multivariable Cox regression analyses for all variables. All statistical analyses were performed using EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), which is a graphical user interface for R.23
Results
Patient characteristics
In this study, DREAM was performed in 137 patients (JMML, n = 124; NS/MPD, n = 13). Characteristics of the 137 patients are summarized in Table 1 and supplemental Table 4. All patients and their 450K (n = 99) and RNA sequencing (n = 78) results have been included in our previous publications.4,7 The median age at diagnosis was 15 months (range, 1-160), and 80 patients (58.4%) showed age-adjusted fetal hemoglobin elevation (supplemental Table 5).24 Of the 137 patients, 90 (66%) underwent allogeneic hematopoietic stem cell transplantation, and the median follow-up period was 33 months (range, 0-291).
DREAM
DREAM8 was performed for 137 samples (discovery cohort, n = 99; validation cohort, n = 38). The median number of sequence reads was 8 269 354 (range, 3 256 572-16 359 479), and the median number of CpG sites with ≥20 reads was 91 515 (range, 31 204-210 378; supplemental Table 6). Results of DREAM of the standard DNA spiked in 19 samples of the discovery cohort are shown in Figure 1D. The median calculated methylation ratios for fully methylated and unmethylated standard DNA were 0.957 (range, 0.841-0.986) and 0.001 (range, 0.000-0.085), respectively. To evaluate the reproducibility of DREAM, a replicate analysis of the 3 independent JMML samples (UPN 7, UPN 101, and UPN 118) was performed. The methylation ratios of CpG sites with ≥20 reads showed good correlation, and the Pearson’s correlation coefficients (r2) of the replicates of UPN 7, UPN 101, and UPN 118 were 0.989, 0.987, and 0.975, respectively (supplemental Figure 1; supplemental Table 7).
Analysis of correlation between methylation ratios of DREAM and β values of 450K array
To investigate how total sequence reads per sample affected the measurement of methylation using the DREAM assay, the sequencing read data of UPN 118 (total, 79 million reads) were randomly reduced into 38 million, 19 million, 9.6 million, 6.3 million, and 3.1 million reads. When all theoretically overlapping 6455 CpG sites were compared, the DREAM methylation ratios were shown to have a significant correlation with Infinium 450K β values, even with 3.1 million reads (supplemental Figure 2; supplemental Table 8).
How the sequencing depth of each CpG site affected the results was also assessed. Moreover, the distribution of the median sequencing depth of 374 921 CpG sites on the genome that could theoretically be analyzed by the DREAM method in 19 cases is also shown (Figure 1E). Pearson’s correlation and Spearman’s rank correlation coefficients between the DREAM methylation ratios and 450K β values of 6455 overlapping CpG sites in 19 samples in the discovery cohort were calculated according to sequence depth. CpG sites with ≥20 reads showed especially good correlation coefficients compared with CpG sites with <20 reads (supplemental Figure 3).
On the basis of these findings, the correlation between the methylation ratios of DREAM and the β values of the 450K platform in 1703 overlapping CpG sites with ≥20 reads in 95% of samples was assessed (supplemental Table 9) using the discovery cohort (n = 99), and a high correlation between the 2 methods was observed (median Pearson’s r2 = 0.954; range, 0.918-0.964; Figure 1F-H; supplemental Figure 4).4
Evaluation of the batch effect of DREAM
The 99 cases in the discovery cohort were subjected to library preparation on 8 different dates, and Pearson’s and Spearman’s correlations between the DREAM methylation ratios and 450K βvalues of 1703 sites were similar according to batch based on library preparation date (supplemental Table 10).
Clustering analysis
Next, an unsupervised hierarchical clustering of the discovery cohort was performed using DREAM data for 7360 promoter-associated CpG sites, and HM (HM_DREAM; n = 35) and LM subgroups (LM_DREAM; n = 64) with a 95% concordance (94 of 99 samples) with the previously reported 450K clustering results were identified4 (Figure 2A). Of the 98 cases that could be compared with the international consensus classification,7 all 90 cases of HM (n = 30) and LM (n = 60) had 100% concordance with the DREAM clustering results. Of the 8 cases classified as the IM group by the international consensus classification, 4 were classified as belonging to the HM_DREAM group and 4 to the LM_DREAM group (Figure 2A).
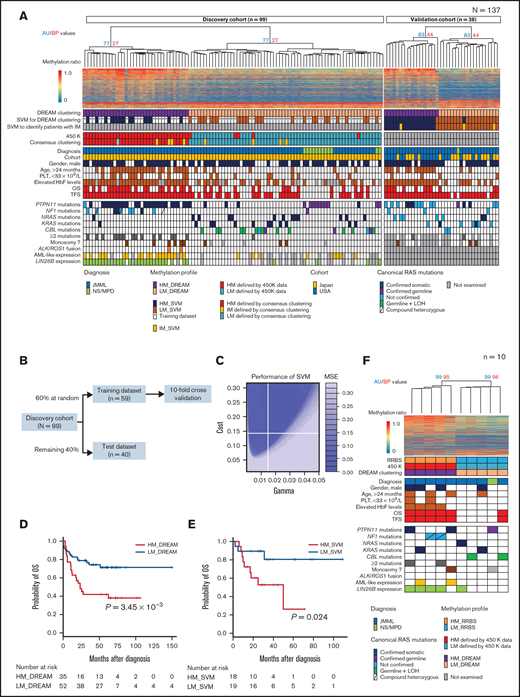
Classifier construction for risk stratification of patients with JMML using DREAM. (A) Patients with JMML or NS/MPD (discovery cohort, n = 99; validation cohort, n = 38) were subjected to unsupervised hierarchical clustering using DREAM data. The heat map displays the methylation ratios calculated for a selected subset of 1000 CpG sites with high differential average methylation levels between HM_DREAM and LM_DREAM subgroups. The methylation ratios were color coded, with a gradual change from blue (0% methylation) to red (100% methylation). Known clinical and biological features were annotated for each patient. Each column indicates 1 patient. (B) Schematic outline of the SVM classifier construction using DREAM data. (C) Performance of SVM. This plot shows the performance of various models using color coding. Darker regions with lower mean square error (MSE) indicate better accuracy. (D) OS of patients with JMML in the discovery cohort using clustering with DREAM data. (E) OS of patients with JMML in the validation cohort using SVM with DREAM data. (F) Patients with JMML (n = 9) and NS/MPD (n = 1) were subjected to unsupervised hierarchical clustering using RRBS data. The heat map displays the methylation ratios calculated for a select 1000 CpG sites with high differential average methylation levels between HM (HM_RRBS) and LM (LM_RRBS) subgroups. HbF, fetal hemoglobin; LOH, loss of heterozygosity; PLT, platelet.
Classifier construction for risk stratification of patients with JMML using DREAM. (A) Patients with JMML or NS/MPD (discovery cohort, n = 99; validation cohort, n = 38) were subjected to unsupervised hierarchical clustering using DREAM data. The heat map displays the methylation ratios calculated for a selected subset of 1000 CpG sites with high differential average methylation levels between HM_DREAM and LM_DREAM subgroups. The methylation ratios were color coded, with a gradual change from blue (0% methylation) to red (100% methylation). Known clinical and biological features were annotated for each patient. Each column indicates 1 patient. (B) Schematic outline of the SVM classifier construction using DREAM data. (C) Performance of SVM. This plot shows the performance of various models using color coding. Darker regions with lower mean square error (MSE) indicate better accuracy. (D) OS of patients with JMML in the discovery cohort using clustering with DREAM data. (E) OS of patients with JMML in the validation cohort using SVM with DREAM data. (F) Patients with JMML (n = 9) and NS/MPD (n = 1) were subjected to unsupervised hierarchical clustering using RRBS data. The heat map displays the methylation ratios calculated for a select 1000 CpG sites with high differential average methylation levels between HM (HM_RRBS) and LM (LM_RRBS) subgroups. HbF, fetal hemoglobin; LOH, loss of heterozygosity; PLT, platelet.
OS and TFS for both cohorts were estimated using the Kaplan-Meier method. Patients with JMML with the HM_DREAM profile in the discovery cohort (excluding those with NS/MPD) had significantly poorer OS and TFS than those with the LM_DREAM profile, with 5-year OS for HM_DREAM of 41.9% (95% CI, 25.3%-57.6%) vs 71.4% (95% CI, 56.2%-82.1%) for LM_DREAM (P = 3.45 × 10−3; Figure 2D). Similarly, 5-year TFS rates for these 2 cohorts were 0% vs 37.4% (95% CI, 23.6%-51.1%), respectively, with a P value of 2.17 × 10−10. On multivariable Cox regression analysis, the HM_DREAM profile was identified as a risk factor in TFS (Table 2).
An unsupervised clustering of the validation cohort (n = 38) was also performed using DREAM data obtained from the aforementioned 7360 promoter-associated CpG sites. Patients in the validation cohort were also classified as belonging to the HM_DREAM (n = 18) or LM_DREAM subgroup (n = 20; Figure 2A).
SVM classifier construction for DREAM clustering
To develop a machine-learning classifier model using an SVM,25 84 CpG sites that were among those exhibiting a distinct difference in average methylation levels (>0.3) between JMML-associated HM_DREAM and LM_DREAM profiles were selected (supplemental Figure 5; supplemental Tables 11-13). Samples from the discovery cohort were randomly assigned to the training (n = 59) or test data set (n = 40) and were used along with the tune.svm function of the e1071 package in R to optimize parameters (Figure 2B).16 The best γ parameter and the best cost parameter were 0.015 and 0.14, respectively. A classifier with the lowest MSE (MSE = 0) was created (Figure 2C).
Using the SVM, patients assigned to the validation cohort were classified as either HM (HM_SVM; n = 18) or LM (LM_SVM; n = 20; Figure 2A). Discrepancies in profiling results between the clustering analysis and the SVM were observed in 2 (5%) of 38 cases in the validation cohort. Patients with the HM_SVM profile had significantly poorer 5-year OS (26.3%; 95% CI, 1.9%-64.0%) than those with the LM_SVM profile (80.5%; 95% CI, 49.1%-93.6%], with a P value of .024 (Figure 2E). On univariable Cox regression analysis, the HM_SVM profile was recognized as a risk factor in OS (supplemental Table 14). No significant differences in TFS were observed, because a majority of patients (33 [86.8%] of 38) in the validation cohort underwent hematopoietic stem cell transplantation (Table 1; supplemental Table 4).
The relationship between the total sequence reads and the number of CpG sites with sufficient sequence depths (≥20 reads) used in the SVM model was evaluated, and 95 (95%) of 99 samples were found to have all 84 CpG sites with ≥20 reads. This was true even for the sample with the lowest number of sequence reads (4 095 131 reads), suggesting that ≥4 million sequence reads was sufficient to classify a patient according to the relevant methylation subgroup (supplemental Figure 6).
SVM classifier construction to identify patients with IM
Among the 7360 sites used for clustering by DREAM, 35 sites that showed a difference of >0.3 in mean methylation value between the consensus IM and the consensus HM were identified. Of the samples in the discovery cohort, those classified as consensus HM/IM (n = 38) were randomly assigned to the training (60% of samples) or test (40% of samples) data set, and these CpG sites were analyzed along with the tune.svm function in the R e1071 package. The SVM_HMIM classifier was created to distinguish between HM and IM (MSE = 0.1; Figure 3A-C; supplemental Table 15). Similarly, 12 sites that showed a difference in methylation values between consensus IM and consensus LM were identified, and the SVM_IMLM classifier was created to distinguish between IM and LM using a sample of consensus IM/LM (n = 68) from the discovery cohort (MSE = 0.05; Figure 3D-F; supplemental Table 16). These SVM classifiers identified 3 patients with putative IM (SVM_IM) from among 38 patients in the validation cohort (Figures 2A and 3G).
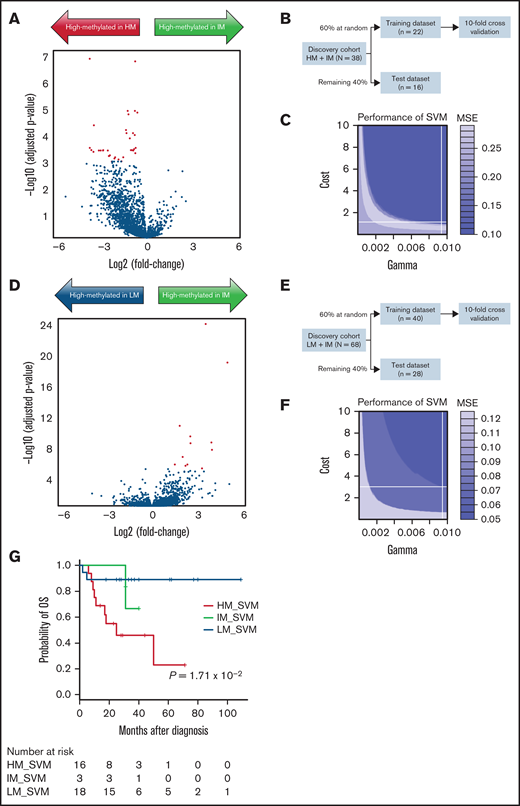
Construction of SVM to identify patients with IM. (A) Using DREAM data of the 38 cases from the discovery cohort that were known to belong to consensus HM (n = 30) and consensus IM (n = 8) subgroups, a volcano plot showing DNA methylation fold changes and P values between consensus HM and IM subgroups was constructed. Red dots indicate 35 CpG sites that showed a distinct difference (>0.3) in mean methylation level. (B) To develop an SVM that classified HM and IM, 60% of the samples were randomly assigned to the training data and the remaining 40% to the test data. (C) In the best SVM model, optimal γ, cost parameter, and MSE were 0.0095, 1.05, and 0.1, respectively. (D) Volcano plot shows DNA methylation fold changes and P values between consensus LM (n = 60) and IM (n = 8) subgroups from the discovery cohort. Red dots indicate 12 CpG sites that showed a distinct difference (>0.3) in mean methylation level. (E) To develop an SVM that classified LM and IM, 60% of the samples were randomly assigned to the training data and the remaining 40% to the test data. (F) In the best SVM model, optimal γ, cost parameter, and MSE were 0.0095, 3.02, and 0.05, respectively. (G) Using these SVM models, 3 of the 38 patients in the validation cohort were classified as belonging to the IM subgroup (IM_SVM). Kaplan-Meier curves for OS were presented using data from 37 cases, excluding 1 case of NS/MPD.
Construction of SVM to identify patients with IM. (A) Using DREAM data of the 38 cases from the discovery cohort that were known to belong to consensus HM (n = 30) and consensus IM (n = 8) subgroups, a volcano plot showing DNA methylation fold changes and P values between consensus HM and IM subgroups was constructed. Red dots indicate 35 CpG sites that showed a distinct difference (>0.3) in mean methylation level. (B) To develop an SVM that classified HM and IM, 60% of the samples were randomly assigned to the training data and the remaining 40% to the test data. (C) In the best SVM model, optimal γ, cost parameter, and MSE were 0.0095, 1.05, and 0.1, respectively. (D) Volcano plot shows DNA methylation fold changes and P values between consensus LM (n = 60) and IM (n = 8) subgroups from the discovery cohort. Red dots indicate 12 CpG sites that showed a distinct difference (>0.3) in mean methylation level. (E) To develop an SVM that classified LM and IM, 60% of the samples were randomly assigned to the training data and the remaining 40% to the test data. (F) In the best SVM model, optimal γ, cost parameter, and MSE were 0.0095, 3.02, and 0.05, respectively. (G) Using these SVM models, 3 of the 38 patients in the validation cohort were classified as belonging to the IM subgroup (IM_SVM). Kaplan-Meier curves for OS were presented using data from 37 cases, excluding 1 case of NS/MPD.
RRBS
Starburst plot and pathway analyses
To integrate DREAM-based DNA methylation data with gene expression data, the relationship between methylation of CpG sites and gene expression by starburst plots was visualized.21 Of the 3564 genes, 77 were significantly downregulated and highly methylated within the HM_DREAM profile as compared with the LM_DREAM profile (Figure 4; supplemental Table 17).
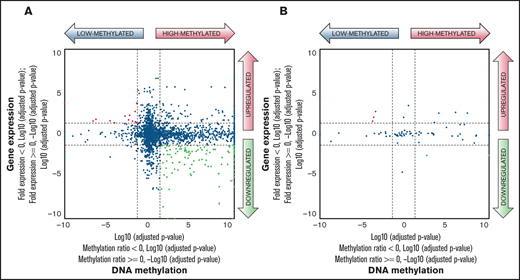
Comparison of DNA methylation and gene expression between HM_DREAM and LM_DREAM profiles. (A) Starburst plot represents the relationship between gene expression levels from transcriptome analysis and DNA methylation levels at 7360 CpG sites used for clustering classification of DREAM among HM_DREAM (n = 32) and LM_DREAM (n = 46) subgroups. Log10 (false discovery rate (FDR)–adjusted P value) was plotted for DNA methylation (x-axis) and gene expression (y-axis) for each gene. In case of a higher mean DNA methylation ratio or mean gene expression value, −1 was multiplied to log10 (FDR-adjusted P value) to provide positive values. The dashed black lines indicate FDR-adjusted P values at 0.05; the red points indicate genes that are significantly upregulated and LM; the green points indicate genes that are significantly downregulated and HM. (B) Starburst plot was evaluated for 84 CpG sites used in the SVM model.
Comparison of DNA methylation and gene expression between HM_DREAM and LM_DREAM profiles. (A) Starburst plot represents the relationship between gene expression levels from transcriptome analysis and DNA methylation levels at 7360 CpG sites used for clustering classification of DREAM among HM_DREAM (n = 32) and LM_DREAM (n = 46) subgroups. Log10 (false discovery rate (FDR)–adjusted P value) was plotted for DNA methylation (x-axis) and gene expression (y-axis) for each gene. In case of a higher mean DNA methylation ratio or mean gene expression value, −1 was multiplied to log10 (FDR-adjusted P value) to provide positive values. The dashed black lines indicate FDR-adjusted P values at 0.05; the red points indicate genes that are significantly upregulated and LM; the green points indicate genes that are significantly downregulated and HM. (B) Starburst plot was evaluated for 84 CpG sites used in the SVM model.
A pathway analysis using DAVID22 was performed for these identified genes. However, the list of enriched genes did not lead to identification of the molecular pathways or gene interaction networks.
Discussion
Outcomes of patients with JMML were successfully predicted by classifying these patients as belonging to HM_DREAM and LM_DREAM subgroups using DREAM, and clear reproducibility of the international consensus classification using the 450K platform was documented,7 despite the fact that a majority of CpG sites used for each classification were not shared (supplemental Figure 7).4,5 The findings were validated in an independent cohort of patients, which again revealed poor prognosis for those with JMML with an HM subgroup profile. Furthermore, a prediction model using SVM as a clinical test was also constructed. To our knowledge, this is the first large-scale study to classify the methylation status of patients with JMML using DREAM, a simple and robust assay.8 The handling time for DREAM is 2 days for preparation of the DREAM libraries (Figure 1A), and it does not require treatment with bisulfite, a factor known to promote sequence errors that can limit the accuracy of quantitation in CpG-rich areas.26 Thus, DREAM is quantitative and highly reproducible, and performing tests on each sample to obtain results in a timely manner is possible. Methylation testing using DREAM in newly diagnosed patients could be used for patient stratification in prospective clinical trials and could be integrated into the clinical decision-making process.
DREAM can theoretically analyze DNA methylation of CpG sites within ∼374 000 SmaI/XmaI recognition sites (5'-CCCGGG-3'), which corresponds to 1.3% of all CpG sites in the human genome.14 In the data of 137 cases in this study, the median number of CpG sites with ≥20 reads was 91 515 (range, 31 204-210 378). DREAM using JMML specimens adequately reproduced the results of the international consensus methylation classification and showed sufficient performance to be used as a clinical test. Furthermore, comparison between DNA methylation and gene expression was visualized in a starburst plot (Figure 4). As in other tumors, a number of genes were identified that showed an inverse correlation between DNA methylation and gene expression levels, but even with various pathway analyses, the biological mechanism by which specific gene methylation is involved in the disease progression of JMML was not fully unraveled.
This study has several limitations. First, DREAM can only assess the methylation level of a relatively small number of CpG sites compared with other next-generation sequencing–based DNA methylation analysis methods, including RRBS and whole-genome bisulfite sequencing. This might be 1 of the reasons why the biological mechanism through which specific gene methylation is involved in the disease progression of JMML has not been identified.
The second limitation is that unsupervised clustering analysis using DREAM methylation data could not separate the IM group, because the proportion of patients classified as IM on the basis of the international consensus classification in the discovery cohort was as low as 8% (8 of 99 patients). An international collaborative study showed that patients with JMML can be divided into 3 groups, HM, IM, and LM, and that most patients with IM have monosomy 7.7 The Japanese cohort had a significantly lower frequency of monosomy 7 compared with the European Working Group cohort (Japan, 9%; European Working Group on Myelodysplastic Syndromes, 22%),7 which may partially explain why IM classification was less frequent only in the Japanese cohort. We recommend that even if patients with monosomy 7 are determined to be LM_DREAM, they should be clinically treated as high-risk cases.
However, a certain number of CpG islands with characteristic methylation levels in the IM group were identified using DREAM methylation data, and the SVM model was constructed, which could separate the IM group (ie, IM_SVM; Figure 3). Using this SVM model, only 3 of the 38 patients in the validation cohort were classified as IM_SVM; therefore, a larger cohort with methylation array data in the future is required to reevaluate whether this SVM model can correctly identify IM patients.
Low platelet count (<33 × 109/L) is a well-established prognostic factor associated with poor prognosis in JMML.27 Low platelet count was significantly associated with both OS and TFS in the validation cohort (supplemental Table 14). In the discovery cohort, it was significantly associated with poor TFS, but not with OS (Table 2). The reason behind this observation is unclear; however, we believe that the relatively small number of transplantations in the discovery cohort and the small number of OS events (37 of 99 cases) did not provide sufficient statistical power.
In conclusion, a simple and robust methylation test (DREAM) that can be incorporated at the time of diagnosis of JMML has been developed. The test can be used to generate a methylation classification for patients, thus allowing them to receive risk-adapted treatment in the context of future clinical trials, such as experimental molecular targeted therapies for the high-risk group or careful follow-up without transplantation for the low-risk group.
Acknowledgments
The authors acknowledge all clinicians, patients, and their families and thank Yoshie Miura, Hiroko Ono, and Chie Amahori for their valuable assistance.
Authorship
Contribution: H.K., K.A., N.M., and M.W. designed and performed the research, analyzed the data, and wrote the paper; E.S. and M.L.L. provided samples and clinical data; K.N., S. Kataoka, D.I., M.H., R.T., K.S., N.K., E.N., A.N., N.N., and A.H. performed the research and analyzed the data; S. Kojima and Y.T. designed the research and wrote the paper; Y.O. and H.M. designed and performed the research, analyzed the data, wrote the paper, and led the entire project and all authors critically reviewed the content of the manuscript and agreed on the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hideki Muramatsu, Department of Pediatrics, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, Japan; e-mail: hideki-muramatsu@med.nagoya-u.ac.jp.