Key Points
Microbial metagenomics were performed on BPDCN skin and bone marrow by using total RNA sequencing, with CTCL and normal skin as controls.
No microbial association with BPDCN was identified, including by a novel computational tool to classify unmapped RNA sequencing reads.

Abstract
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is a hematologic malignancy believed to originate from plasmacytoid dendritic cells (pDCs), the immune cells responsible for producing type 1 interferons during infection. Nearly all patients with BPDCN have prominent skin involvement, with cutaneous infiltration occupying the dermis and subcutis. One half of patients present with BPDCN cells only in the skin, with no evidence of disease elsewhere. Because normal pDCs are rare or absent in cutaneous sites, and they only traffic to the skin after activation by pathogen or inflammation, our aim was to determine if a microorganism is associated with BPDCN. We performed RNA sequencing in BPDCN skin and bone marrow, with cutaneous T-cell lymphoma (CTCL) and normal skin as controls. GATK-PathSeq was used to identify known microbial sequences. Bacterial reads in BPDCN skin were components of normal flora and did not distinguish BPDCN from controls. We then developed a new computational tool, virID (Viral Identification and Discovery; https://github.com/jnoms/virID), for identification of microbial-associated reads remaining unassigned after GATK-PathSeq. We found no evidence for a known or novel virus in BPDCN skin or bone marrow, despite confirming that virID could identify Merkel cell polyomavirus in Merkel cell carcinoma, human papillomavirus in head and neck squamous cell carcinoma, and Kaposi’s sarcoma herpesvirus in Kaposi’s sarcoma in a blinded fashion. Thus, at the level of sensitivity used here, we found no clear pathogen linked to BPDCN.
Introduction
Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is an orphan hematologic malignancy with poor survival.1 It has several unique properties. First, although BPDCN is a hematologic cancer, 90% of patients have skin involvement and 50% have disease detectable only in the skin at presentation. Second, skin lesions can persist despite complete responses at other sites such as bone marrow.2 Third, the hypothesized cell of origin is the plasmacytoid dendritic cell (pDC), the principal producer of type 1 interferons in response to viral pathogens.3 We postulated that a skin-resident pathogen might drive BPDCN and contribute to these clinical characteristics. Here, we report an unbiased metagenomic analysis of microbial associations with BPDCN.
Methods
Samples and RNA sequencing
Patients were consented to an institutional review board–approved protocol. BPDCN (n = 5) and cutaneous T-cell lymphoma (CTCL) mycosis fungoides type (n = 5) samples were collected via punch biopsy. Bone marrow was also collected from patients with BPDCN. Additional clinical information, including age, sex, biopsy site, immunophenotypic markers, and extent of disease involvement, are given in supplemental Table 1. Normal skin samples (n = 5) were obtained from individuals who had undergone complete resection of basal cell carcinoma, and at the time of repair, normal tissue was taken from sites farthest from the negative surgical margin. All biopsy specimens were freshly frozen (ie, not fixed).
The Qiagen RNeasy Mini kit was used to extract RNA from fresh-frozen biopsy specimens. Total RNA libraries were prepared by using the NEBNext Ultra II Directional RNA Library Prep kit (New England Biolabs) and were sequenced on a HiSeq 2500 (Illumina).
Normal skin, BPDCN skin, BPDCN bone marrow, and CTCL skin sequences are available at the Sequence Read Archive (accession PRJNA596800). Merkel cell carcinoma transcriptome sequences (n = 6) were previously described.4 Head and neck squamous cell carcinoma RNA sequencing (RNA-seq) data (n = 10) were accessed through The Cancer Genome Atlas.5 Sample IDs were human papillomavirus (HPV)–16 positive (n = 5), HNSC-BA-A4IH-TP, HNSC-BB-7866-TP, HNSC-CN-A499-TP, HNSC-CR-7404-TP, and HNSC-HD-A634-TP; and HPV-16 negative (n = 5), HNSC-4P-AA8J-TP, HNSC-BA-4074-TP, HNSC-BA-4075-TP, HNSC-BA-4076-TP, and HNSC-BA-5151-TP. Kaposi’s sarcoma lesions (n = 4) and contralateral normal skin (n = 4) RNA-seq were previously described.6
Host gene expression analysis
Transcript abundance was quantified by using kallisto,7 and differentially expressed genes were identified with sleuth8 using R 3.5.2 (R Foundation for Statistical Computing),9 selected according to a Benjamini-Hochberg–corrected q value ≤0.05. Samples were clustered by using Ward’s minimum variance method based on Euclidean distance.
Metagenomic analysis
A stepwise approach was implemented for metagenomic classification of RNA-seq reads. First, GATK-PathSeq10 was used to computationally subtract reads that mapped to the GATK-PathSeq host reference. GATK-PathSeq then mapped remaining reads against a comprehensive microbial reference using BWA-MEM.11
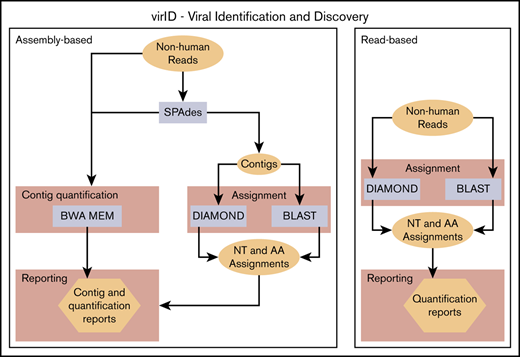
We developed a novel metagenomic analysis pipeline, virID (Viral Identification and Discovery), to assign reads that remained unclassified after GATK-PathSeq. This pipeline implements 2 approaches. In assembly-based virID, reads are de novo assembled into longer sequences (contigs) by using rnaSPAdes.12 Next, contigs are taxonomically assigned with MegaBLAST13 and DIAMOND.14 These algorithms are more sensitive to divergent sequences than GATK-PathSeq’s implementation of BWA-MEM owing to more extensive reference databases and in DIAMOND’s case, the use of an amino acid rather than nucleotide reference. MegaBLAST aligns contigs with the NCBI nucleotide “nt” reference database, and DIAMOND translates each contig into amino acid sequences in all 6 reading frames and searches these against the RefSeq protein database. Although DIAMOND is generally more sensitive to divergent sequences than MegaBLAST due to its translated amino acid search, MegaBLAST is an effective measure to control for false-positive assignments coming from DIAMOND. Each contig is then assigned to the last common ancestor of its top matches, and the MegaBLAST and DIAMOND results are merged. Raw reads are mapped back to each contig using BWA-MEM for quantification. To increase the likelihood of sequence identification in samples with poor assembly efficiency, we also implemented a virID read-based approach in which GATK-PathSeq–unassigned reads were profiled directly with MegaBLAST and DIAMOND.
Finally, we implemented a reference-independent “kmer enrichment” strategy to identify reads from nonrepetitive sequences enriched in BPDCN skin. In this approach, all 21 bp sequences (21mers) present in at least 2 BPDCN but no control skin were collected; reads containing these 21mers were identified and taxonomically assigned with BLASTn.
virID and scripts used for kmer enrichment are available at https://github.com/jnoms/virID.
Results and discussion
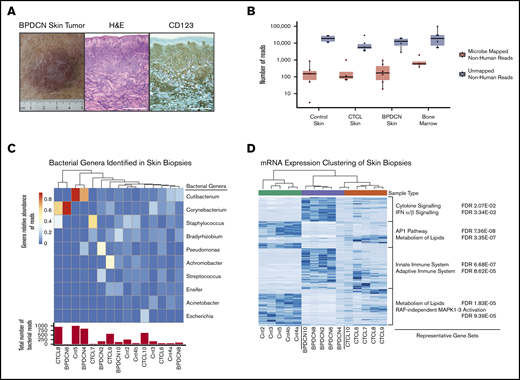
Specific pathogens are associated with some skin-localized malignancies, such as Kaposi’s sarcoma–associated herpesvirus in Kaposi’s sarcoma15 and Merkel cell polyomavirus (MCPyV) in Merkel cell carcinoma.16 Given the role of pDCs in microbial sensing, particularly for viruses, we sought to determine if there is a microorganism associated with BPDCN. We performed RNA-seq of BPDCN skin tumors and paired bone marrow aspirates from the same patient at the same time point. Bone marrow was analyzed to determine if any identified microorganism was present only in skin or if it was also associated with BPDCN at other sites. To determine if any identified organisms were specific to BPDCN, we also sequenced CTCL and normal skin biopsy samples. Pathology was confirmed in all cases (Figure 1A).
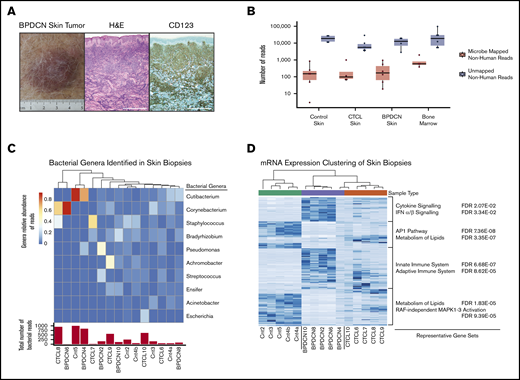
GATK-PathSeq analysis of BPDCN skin transcriptomes reveals normal cutaneous microbiota. (A) BPDCN skin tumor and biopsy stained with hematoxylin and eosin (H&E) and for CD123, representative of all BPDCNs sampled containing similar relative amount of tumor. Scale bars, 0.5 mm. (B) The number of GATK-PathSeq non-host microbe-mapped reads and non-host unmapped reads. (C) (Top) Genera relative abundance of GATK-PathSeq bacteria-assigned reads in skin samples. For clarity, only the top 10 genera are displayed. Samples are clustered with Euclidean distance based on the relative abundance of these 10 genera. (Bottom) Absolute number of GATK-PathSeq mapped reads assigned to the superkingdom bacteria in each sample (Cnt = normal skin). Color bar is genera relative abundance of reads. (D) Unsupervised hierarchical clustering using the 315 most differentially expressed human genes (rows) in the 15 skin samples (columns). Genes associated with each cluster were tested for enrichment in the c2.cp (canonical pathways) gene set deposited in the Molecular Signatures Database v7.0, and representative gene sets are displayed. FDR, false discovery rate; IFN, interferon; mRNA, messenger RNA.
GATK-PathSeq analysis of BPDCN skin transcriptomes reveals normal cutaneous microbiota. (A) BPDCN skin tumor and biopsy stained with hematoxylin and eosin (H&E) and for CD123, representative of all BPDCNs sampled containing similar relative amount of tumor. Scale bars, 0.5 mm. (B) The number of GATK-PathSeq non-host microbe-mapped reads and non-host unmapped reads. (C) (Top) Genera relative abundance of GATK-PathSeq bacteria-assigned reads in skin samples. For clarity, only the top 10 genera are displayed. Samples are clustered with Euclidean distance based on the relative abundance of these 10 genera. (Bottom) Absolute number of GATK-PathSeq mapped reads assigned to the superkingdom bacteria in each sample (Cnt = normal skin). Color bar is genera relative abundance of reads. (D) Unsupervised hierarchical clustering using the 315 most differentially expressed human genes (rows) in the 15 skin samples (columns). Genes associated with each cluster were tested for enrichment in the c2.cp (canonical pathways) gene set deposited in the Molecular Signatures Database v7.0, and representative gene sets are displayed. FDR, false discovery rate; IFN, interferon; mRNA, messenger RNA.
Our strategy was to iteratively classify sequencing reads. First, we used GATK-PathSeq to subtract host (human) reads and query resultant reads against a microbial reference. On average, only ∼0.01% of total reads were nonhuman, with ∼0.0005% assigned to known microorganisms (Figure 1B; supplemental Figure 1A). Most of these reads were bacterial (supplemental Figure 1B). The skin samples included bacterial genera that are considered normal skin flora, including Cutibacterium, Corynebacterium, and Staphylococcus (Figure 1C).17 Hierarchical clustering by relative abundance of reads in a given bacterial genera did not group the samples according to tumor type, in contrast to clustering based on human gene expression that clearly did (Figure 1D). These data suggest that no distinct bacterial community defines BPDCN in the skin. We did not control directly for bacterial RNA extraction efficiency, and thus it remains possible that an important bacterial population could be identified in future studies.
GATK-PathSeq did not identify reads mapping to a known virus in any skin sample. Although some studies have reported viruses such as HTLV-1, Epstein-Barr virus, or protoparvoviruses associated with a minority of CTCLs, there has been no infectious agent consistently linked to the disease to date.18,19 In agreement with those data, no viruses were detected in the 5 CTCLs we investigated here.
Ten- to 100-fold more unassigned reads than microbe-assigned reads remained after the GATK-PathSeq analysis. To address the possibility that a known or novel virus was present in the unassigned reads, we designed a custom bioinformatics pipeline, virID (Figure 2A). The purpose of virID is to perform unbiased mapping and identification of reads by querying assembled sequences against microbial reference sequences.
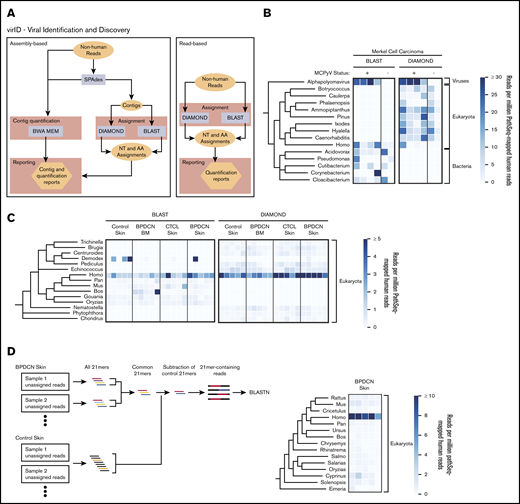
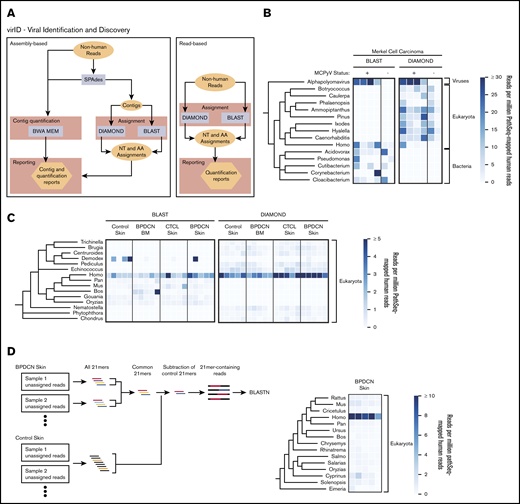
virID, a novel microbial sequence identification algorithm, identifies no known or novel viral sequences associated with BPDCN. (A) Schematic of the virID computational pipeline. virID can be run with assembly (left) or without assembly (right). In the assembly-based approach, SPAdes is used to conduct de novo assembly of GATK-PathSeq unassigned reads to generate contigs. Contigs are then subjected to nucleotide (MegaBLAST) and translated amino acid (DIAMOND) searches against reference databases. The number of reads supporting each contig is determined by mapping reads back to contigs with the BWA-MEM aligner. Results are then integrated to report the abundance of microorganisms in the input reads. In the read-based approach, reads are directly subjected to BLAST and DIAMOND searches. (B) Taxonomic representation of assembly-based assignment when applied to 4 virus-positive and 2 virus-negative Merkel cell carcinomas. MCPyV was excluded from the virID reference databases. The top 15 genera per mean abundance are displayed. The tree is taxonomic and does not incorporate phylogenetic distances. Units are genera reads per 1 million human reads. (C) Taxonomic representation of read-based assignment of GATK-PathSeq unmapped reads. The top 15 genera by mean abundance are displayed. Units are genera reads per 1 million human reads. (D, left) Schematic of the kmer-enrichment approach. First, all 21mers were identified in the unassigned reads from BPDCN skin and control skin samples using jellyfish.22 21mers present in at least 2 BPDCN samples were kept, and any 21mer present in any control sample was removed. Reads containing remaining 21mers were subjected to BLASTN homology search. (D, right) Results from BLASTN search of kmer-enriched reads. The top 15 genera by mean abundance are displayed. Units are genera reads per 1 million human reads.
virID, a novel microbial sequence identification algorithm, identifies no known or novel viral sequences associated with BPDCN. (A) Schematic of the virID computational pipeline. virID can be run with assembly (left) or without assembly (right). In the assembly-based approach, SPAdes is used to conduct de novo assembly of GATK-PathSeq unassigned reads to generate contigs. Contigs are then subjected to nucleotide (MegaBLAST) and translated amino acid (DIAMOND) searches against reference databases. The number of reads supporting each contig is determined by mapping reads back to contigs with the BWA-MEM aligner. Results are then integrated to report the abundance of microorganisms in the input reads. In the read-based approach, reads are directly subjected to BLAST and DIAMOND searches. (B) Taxonomic representation of assembly-based assignment when applied to 4 virus-positive and 2 virus-negative Merkel cell carcinomas. MCPyV was excluded from the virID reference databases. The top 15 genera per mean abundance are displayed. The tree is taxonomic and does not incorporate phylogenetic distances. Units are genera reads per 1 million human reads. (C) Taxonomic representation of read-based assignment of GATK-PathSeq unmapped reads. The top 15 genera by mean abundance are displayed. Units are genera reads per 1 million human reads. (D, left) Schematic of the kmer-enrichment approach. First, all 21mers were identified in the unassigned reads from BPDCN skin and control skin samples using jellyfish.22 21mers present in at least 2 BPDCN samples were kept, and any 21mer present in any control sample was removed. Reads containing remaining 21mers were subjected to BLASTN homology search. (D, right) Results from BLASTN search of kmer-enriched reads. The top 15 genera by mean abundance are displayed. Units are genera reads per 1 million human reads.
For validation, we implemented virID on skin RNA-seq from 6 Merkel cell carcinoma tumors after host subtraction with GATK-PathSeq. Four of these tumors were known to contain MCPyV, an alphapolyomavirus, and 2 did not. virID identified clear enrichment of the genus Alphapolyomavirus in the 4 virus-positive samples even when MCPyV was excluded from the virID reference database (Figure 2B). As additional validation, we implemented virID on publicly available RNA-seq data from 2 other virus-associated cancers after removal of the etiologic virus from the virID reference databases. virID identified the family Papillomaviridae in 5 HPV-16–positive (but not 5 HPV-16–negative) head and neck squamous cell carcinoma samples available through The Cancer Genome Atlas (supplemental Figure 2A). Similarly, virID detected the family Herpesviridae in 4 Kaposi’s sarcoma lesions but not in contralateral non-cancer tissue from each patient (supplemental Figure 2B). In these 2 cohorts, DIAMOND assigned reads or contigs to viruses in the same family as the known etiologic virus.
virID applied to BPDCN skin and bone marrow, and to CTCL and normal skin, showed no clear evidence of a known or novel virus associated with either malignancy (supplemental Figure 3A). However, assembly efficiency was poor, including failed assembly for 5 samples, possibly related to the low number of total non-human reads. We therefore implemented virID without the assembly step, querying reads only, which still failed to identify a biologically relevant organism in the tumor samples (Figure 2C). Both assembly and read-based approaches identified a human Papillomavirus species in 1 BPDCN sample, which can be a normal component of the skin virome.20 This single identified HPV is most similar to HPV-mSK_224, an isolate that was first identified on the skin of a patient with a primary immunodeficiency (DOCK8 deficiency).21 Phylogenetic analysis of the HPV-mSK_224 L1 sequence suggests this virus is most similar to HPV-161, a low-risk gammapapillomavirus (supplemental Figure 3B).
Even after using virID, up to 90% of the non-human reads remained unclassified. RepeatMasker revealed that up to 50% of these unclassified reads included repetitive human ribosomal gene sequences (supplemental Figure 3C). To interrogate the remaining reads, we used a reference-independent approach to identify recurrent 21mers present in BPDCN but not in control skin samples. We reasoned that if an as-yet completely unknown microbe was associated with a majority of BPDCNs, there would be shared contiguous sequences among disease biopsy samples but not in controls. BLASTn search of reads containing enriched 21mers failed to identify a potential pathogen (Figure 2D).
In conclusion, we implemented a variety of bioinformatics approaches to characterize the metagenome of BPDCN skin tumors and found no microorganism uniformly associated with the disease. Our results indicate that BPDCN does not have a viral driver. However, it remains possible that a microorganism is relevant earlier in BPDCN ontogeny, in a tissue or subset of patients not analyzed here, or that our approach lacked sufficient sensitivity. Further studies are therefore necessary to understand mechanisms driving the unique skin tropism and clinical characteristics of BPDCN.
Normal skin, BPDCN skin, BPDCN bone marrow, and CTCL skin RNA sequences are available at the Sequence Read Archive (accession PRJNA596800).
Acknowledgments
The authors thank Mingjie Wang and Ami Bhatt for helpful discussions.
This work was supported by the National Cancer Institute, National Institutes of Health (grant CA225191-01) (A.A.L.), and by the Dana-Farber Medical Oncology Translational Research Program (A.A.L.). Portions of this research were conducted by using the O2 High Performance Compute Cluster, supported by the Research Computing Group at Harvard Medical School.
Authorship
Contribution: All authors collected samples, generated data, analyzed data, and edited the manuscript; and J.N. and A.A.L. designed the study and wrote the manuscript.
Conflict-of-interest disclosure: A.A.L. receives research support from AbbVie and Stemline Therapeutics; and is a consultant for N-of-One/Qiagen. M.M. receives research support from Bayer, Ono, and Janssen; has patents licensed to Bayer and LabCorp; and is a consultant for OrigiMed. J.A.D. receives research support from Constellation Pharmaceuticals; and is a consultant to EMD Serono, Inc. and to Merck & Co. Inc. The remaining authors declare no competing financial interests
Correspondence: Andrew A. Lane, Dana-Farber Cancer Institute, 450 Brookline Ave, Mayer 413, Boston, MA 02215; e-mail: andrew_lane@dfci.harvard.edu.