Key Points
Low variant allele frequency clonal hematopoiesis or aberrant CD7 expression on hematopoietic stem cells increases the risk of tMN.
Patients at high risk of developing tMN after ASCT can be identified at the time of leukapheresis.

Abstract
Therapy-related myeloid neoplasms (tMN) develop after exposure to cytotoxic and radiation therapy, and due to their adverse prognosis, it is of paramount interest to identify patients at high risk. The presence of clonal hematopoiesis has been shown to increase the risk of developing tMN. The value of analyzing hematopoietic stem cells harvested at leukapheresis before autologous stem cell transplantation (ASCT) with next-generation sequencing and immunophenotyping represents potentially informative parameters that have yet to be discovered. We performed a nested case-control study to elucidate the association between clonal hematopoiesis, mobilization potential, and aberrant immunophenotype in leukapheresis products with the development of tMN after ASCT. A total of 36 patients with nonmyeloid disease who were diagnosed with tMN after treatment with ASCT were included as case subjects. Case subjects were identified from a cohort of 1130 patients treated with ASCT and matched with 36 control subjects who did not develop tMN after ASCT. Case subjects were significantly poorer mobilizers of CD34+ cells at leukapheresis (P = .016), indicating that these patients possess inferior bone marrow function. Both clonal hematopoiesis (odds ratio, 5.9; 95% confidence interval, 1.8-19.1; P = .003) and aberrant expression of CD7 (odds ratio, 6.6; 95% confidence interval, 1.6-26.2; P = .004) at the time of ASCT were associated with an increased risk of developing tMN after ASCT. In conclusion, clonal hematopoiesis, present at low variant allele frequencies, and aberrant CD7 expression on stem cells in leukapheresis products from patients with nonmyeloid hematologic cancer hold potential for the early identification of patients at high risk of developing tMN after ASCT.
Introduction
Therapy-related myeloid neoplasms (tMN) are high-risk neoplasms evolving after exposure to a number of antineoplastic agents, including alkylating agents, topoisomerase inhibitors, and radiotherapy; they include therapy-related myeloproliferative neoplasms (tMPN), myelodysplastic syndrome (tMDS), and acute myeloid leukemia (tAML).1 As such, cytoreduction as part of autologous stem cell transplantation (ASCT) increases the risk of developing tMN.2,3 Given the increasing use of ASCT, better supportive care, and new and more potent antineoplastic drugs available for several lines of therapy, the incidence of tMN is expected to further increase in the future.4 As reflected in the high frequency of adverse cytogenetic and mutational profiles in this subset of patients, it is well established that patients diagnosed with tMN experience a dismal prognosis.5-9
Clonal hematopoiesis of indeterminate potential (CHIP) constitutes a premalignant condition that is characterized by somatic mutations with a variant allele frequency (VAF) ≥0.02 in genes related to hematologic malignancies in patients with absence of cytopenia. However, no meaningful biological argument for a VAF limit of 0.02 has ever been proposed.10 The term clonal hematopoiesis (CH) more widely describes detectable somatic mutations at any VAF and thus contains the entire spectrum of premalignant conditions related to somatic mutations in genes associated with myeloid disorders. CH has been shown to increase all-cause mortality, atherosclerosis, and the risk of developing de novo hematologic neoplasms, as well as tMN.11-16 Moreover, in patients with CH, high-dose conditioning regimens as part of treatment with ASCT have been shown to provide a proliferative advantage for mutated cell clones.17 The impact of individual mutations in malignant transformation is still being investigated. However, it has become evident that mutations in both DNMT3A and TET2 can be detected in a large fraction of the healthy adult population18 ; individually, these clones often display little to no clonal evolution over time and are associated with a lower increase in risk of developing myeloid neoplasms compared with mutations in TP53, IDH1/2, and spliceosome genes.19-22
The prevalence of CH increases with age, with an estimated 10% of the general population expected to exhibit CH by 70 years of age.23-25 Of note, CH has been shown to originate in the hematopoietic stem cell or in a common progenitor.26 In myeloid malignancies, accumulating evidence suggests that the neoplastic cell population is maintained by rare and distinct subsets of cells with stem cell features, so-called leukemic stem cells (LSCs). Identification of LSCs is primarily based on immunophenotyping by using flow cytometry (FCM). A number of cell surface markers have been shown to be aberrantly expressed on the CD34+CD38– stem cells and progenitor cells in AML.27-29 Several markers have been proposed as useful in the effort to identify putative LSCs, including the C-type lectin domain family 12 member A receptor (CLEC12A), CD7, CD19, and CD123, all of which have been shown to be aberrantly expressed in the CD34+CD38– subset in patients with AML.27-32 The presence of either CH or putative LSCs may drive initiation of tMN in patients who have been intensively treated for a nonmyeloid hematologic neoplasm, with the intent to either cure or achieve a long-lasting remission.
Thus, in the current study, we investigated the presence of CH and LSCs in patients’ leukapheresis products before ASCT, as well as their potential to predict development of tMN. We hypothesized that patients with nonmyeloid primary hematologic neoplasms undergoing ASCT, who developed tMN, had detectable low VAF CH at time of leukapheresis, and that these mutations represented a risk factor for the development of tMN. We further investigated if minute fractions of LSCs were present in leukapheresis products and whether their identification could be a useful predictor of developing tMN.
Materials and methods
Patients
The cohort of this nested case-control study consists of patients treated with ASCT at the Department of Hematology, Aarhus University Hospital (Aarhus, Denmark), between 1989 and 2016. Cases were identified via the Danish Pathology Registry, and only patients without any signs of myeloid disease, clinically or in the bone marrow biopsy results before ASCT, were included. All tMN diagnoses were carefully reviewed and verified by an experienced pathologist. Case subjects were patients with nonmyeloid primary disease, diagnosed with tMN after ASCT (minimum latency being 90 days). Control subjects were patients with nonmyeloid primary disease who did not develop tMN after ASCT. Matching 1:1 was performed on the following variables listed in order of priority: (1) follow-up time (defined as time elapsed between ASCT and tMN diagnosis for cases); (2) age (±5 years); (3) sex; and (4) conditioning regimen. Criteria for inclusion for both case and control subjects were: (1) age 18 to 75 years; (2) treatment with ASCT at the Department of Hematology at Aarhus University Hospital; and (3) availability of biobanked mononuclear cells obtained from leukapheresis before ASCT. Criteria for exclusion were: (1) treatment of tMN at time of ASCT; (2) myeloid disease as indication for ASCT; and (3) patients who opposed the experimental use of stored tissue as registered in the national “Tissue Application Register.” A total of 36 cases were included in the study (Figure 1).
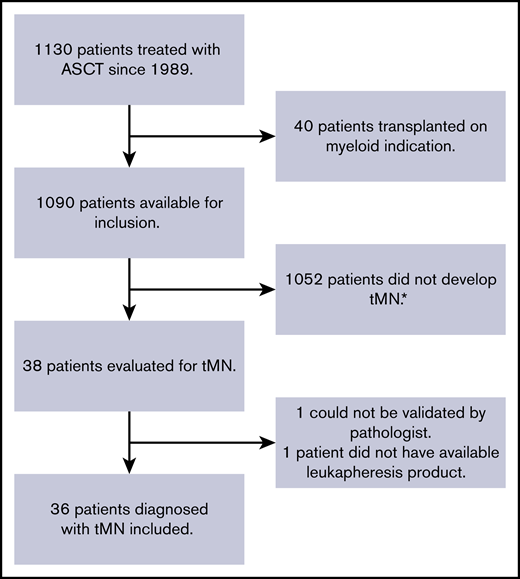
Flowchart describing inclusion of tMN patients in the case cohort. The cohort consisted of patients treated with ASCT at the Department of Hematology, Aarhus University Hospital, from 1989 to 2016. *Control subjects selected from this group.
Flowchart describing inclusion of tMN patients in the case cohort. The cohort consisted of patients treated with ASCT at the Department of Hematology, Aarhus University Hospital, from 1989 to 2016. *Control subjects selected from this group.
The study was approved by the Central Denmark Region Committees on Health Research Ethics (record no. 1-10-72-396-17) and the Danish Data Protection Agency (record no. 1-16-02-851-17). The project was conducted in accordance with the Declaration of Helsinki.
Flow cytometry
Mononuclear cells harvested as part of leukapheresis before ASCT were stored in liquid nitrogen in a dimethyl sulfoxide solution. The cryopreserved mononuclear cells were thawed in 37°C water bath and resuspended in a thawing buffer containing 10% heat-inactivated fetal calf serum (Biochrom, Berlin, Germany). Cells were stained with the following pretitrated monoclonal antibodies: CD123 FITC (clone AC145; Miltenyi, Bergisch Gladbach, Germany), CLEC12A PE (clone HB3; Department of Hematology, Aarhus University Hospital), CD14 ECD (clone RMO52; Beckman Coulter, Brea, CA), CD34 PerCP-Cy5.5 (clone 581; BioLegend, San Diego, CA), CD117 PE-Cy7 (clone 104D2; BD Biosciences, Franklin Lakes, NJ), CD7 APC (clone 124-1D1; Thermo Fisher Scientific, Waltham, MA), CD19 APC-A700 (clone J3-119; Beckman Coulter), CD38 APC-H7 (clone HB7; BD Biosciences), HLA-DR PB (clone L243; BioLegend), and CD45 KrO (cloneJ.33; Beckman Coulter). Data were acquired on a Navios flow cytometer (Beckman Coulter [10 colors; 405, 488, and 638 nm laser; 5+3+2 photomultiplier tube configuration]) using the Navios Tetra acquisition software. Compensation was set by using UltraComp eBeads (Thermo Fisher Scientific). Data were analyzed by using Kaluza Analysis software, version 1.3 (Beckman Coulter) and illustrated by using FlowJo Data Analysis software, version 10.6.1 (BD Biosciences).
Analysis was performed independently by 2 experienced investigators, and both were blinded to case/control status. The gating strategy is described in detail in supplemental Figure 1. Erythroid cells (CD45neg/SSClow) and lymphocytes (CD45high/SSClow) were used as the internal negative/positive gating control for aberrant antigen expression. A median number of 118 CD34+CD38– cells (range, 6-1281) were analyzed. The difference in absolute numbers of CD34+CD38– cells did not reach statistical significance. A minimum of 5 clustered cells was regarded as a positive lower limit in the statistical analysis.
Targeted deep sequencing
Samples were subjected to targeted next-generation sequencing (NGS) using the commercially available Myeloid Tumor Solution targeted panel (SOPHiA Genetics, Saint Sulpice, Switzerland). NGS was performed in accordance with the manufacturer’s instructions and subsequently analyzed in SOPHiA DDM analysis software (SOPHiA Genetics). The panel covers 30 genes recurrently mutated in myeloid neoplasms: ABL, ASXL1, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NPM1, NRAS, PTPN11, RUNX1, SETBP1, SF3B1, SRSF2, TET2, TP53, U2AF1, WT1, and ZRSR2. Cutoffs for VAF and read depth for the variant calling algorithm was determined by analysis of variant call format (.vcf) files from all patients (supplemental Figure 2). Variants were excluded if: (1) read depth was <3000 reads; (2) VAF was <0.003; (3) variant location was outside of ±25 nucleotides of coding regions; (4) variant was an indel present in a homopolymeric stretch; and (5) variant was suspected germline with VAF over 0.95, or between 0.45 and 0.55, and reported in the Exome Aggregation Consortium database.33 Mutations in DNMT3A and TET2 were excluded from the statistical analysis because they have been frequently reported as benign age-related changes in the hematopoietic stem cells; they are shown to be frequently present in the healthy population and reportedly confer a low risk of malignant transformation.18,19,21,34 Data from the NGS analysis were independently evaluated by 2 investigators.
Statistical analyses
Continuous variables were compared by using the Wilcoxon rank sum nonparametric test. Categorical variables were analyzed by using the Pearson χ2 test. All estimates were reported with a 95% confidence interval (CI), and P < .05 was considered significant. Stata version 15.1 (StataCorp LLC, TX) was used for statistical analyses.
Results
Study cohort
The cohort consisted of 36 tMN patients and 36 control subjects. Of the 36 cases, 10 patients were diagnosed with tAML, 25 with tMDS, and 1 with unclassifiable tMPN. In total, 7 (19.4%) of 36 patients were female, and the median age at ASCT was 55 years (range, 29-69 years) for patients and 56 years (range, 35-67 years) for control subjects (Table 1). All patients received an alkylating agent, a topoisomerase II inhibitor, or both as part of their conditioning regimen. No difference was observed in the proportion of patients treated with cytotoxic maintenance therapy post-ASCT between the groups (supplemental Table 1A-B). Median time from ASCT to diagnosis of tMN was 3.5 years (range, 0.2-18.1 years), and patients with tMDS and tAML presented with similar clinical characteristics at diagnosis (supplemental Table 2).
Stem cell mobilization at leukapheresis
As part of the leukapheresis, all patients were stimulated with granulocyte colony-stimulating factor and a priming regimen (supplemental Table 1A-B). Case subjects mobilized significantly fewer CD34+ stem cells than control subjects (Table 1), with a median of 3.57 × 106 cells/kg (range, 0.4-29 × 106 cells/kg) compared with 5.32 × 106 cells/kg (range, 2-18 × 106 cells/kg; P = .016), indicating that the normal hematopoiesis could be impaired as early as at time of ASCT. No differences were found in regeneration time of neutrophilic granulocytes or platelets, or in the time of admission.
Clonal hematopoiesis at ASCT and risk of tMN
Targeted NGS of case and control subjects was performed to reveal the presence of CH (supplemental Table 3). Low VAF mutations in DNMT3A and TET2 were detected at an equal rate in both case subjects (50% DNMT3A, 16% TET2) and control subjects (33% DNMT3A, 28% TET2), and they did not correlate to an increased risk of developing tMN (P = .216) (supplemental Table 4). After exclusion of DNMT3A and TET2 from the statistical analysis, the most frequently mutated genes in patients with tMN were TP53 (26%), ASXL1 (20%), and ZRSR2 (13%) (Figure 2A). The predicted effects of mutations detected were mainly missense, with mutations resulting in frameshifts, stop-codons, and splice-donor mutations being exclusive to case subjects (supplemental Figure 3A,C). The single nucleotide base shift T>C/G>A was the most frequently detected and represented 32% (12 of 37) of the total mutations. Base substitutions, C>T/A>G, G>C and A>C/G>T, were exclusive to case subjects (supplemental Figure 3B). Mutations were distributed evenly between age groups, and, remarkably, all patients harboring mutations at ASCT who were aged <50 years developed tMN (Figure 2B). In cases, 47% (95% CI, 30-64) had one or more mutations at time of ASCT compared with 14% (95% CI, 4-29) of control subjects. Detectable CH at time of ASCT was significantly associated with developing tMN, corresponding to a factor 5.9 increase in risk (OR, 5.9; 95% CI, 1.8-19.1; P = .003) (supplemental Table 5). In one patient (Figure 2C), NGS analysis was also performed at tMN diagnosis. This analysis revealed the clonal expansion of a mutation in the spliceosome gene (SRSF2) from VAF 0.005 at ASCT to VAF 0.45 at tMDS. Of the other mutations detected at ASCT, none persisted at time of tMN, while other mutations had emerged (data not shown).
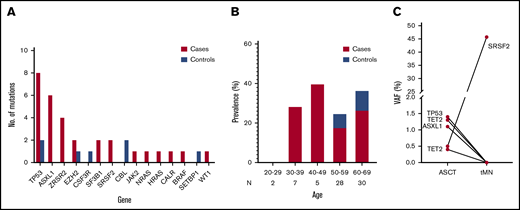
Somatic mutations detected pre-ASCT. Results of targeted deep sequencing of case (red) and control subjects (blue). (A) Total number of mutations according to gene in case and control subjects. (B) Frequency of patients testing positive for one or more mutation(s) in each age group. (C) NGS analysis of the mutational profile of patient 1185 at time points ASCT and tMN diagnosis. N, total number of patients in each age group.
Somatic mutations detected pre-ASCT. Results of targeted deep sequencing of case (red) and control subjects (blue). (A) Total number of mutations according to gene in case and control subjects. (B) Frequency of patients testing positive for one or more mutation(s) in each age group. (C) NGS analysis of the mutational profile of patient 1185 at time points ASCT and tMN diagnosis. N, total number of patients in each age group.
Evaluation of LSCs
Analysis of aberrant marker expression on CD34+CD38– stem cells (Table 1; Figure 3) revealed a significant difference in the presence of aberrant CD7+ stem cells between case and control subjects, resulting in a factor 6.6 increase in risk of developing tMN after ASCT (OR, 6.6; 95% CI, 1.6-26.2; P = .004) (supplemental Table 5). Evaluation of aberrant expression of CLEC12A, CD19, and CD123 on CD34+CD38– stem cells revealed no differences between case and control subjects.
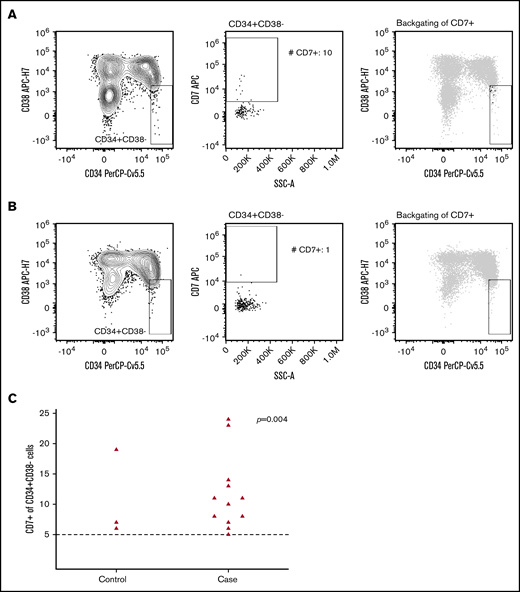
Flow cytometric analysis of aberrant CD7 expression within the stem cell subset. The CD34+CD38– stem cells were identified within the CD45lowSSClowCD14– cells (left panel), and the presence (A) or absence (B) of aberrant CD7+ cells were evaluated by using a bivariate plot of SSC-A vs CD7 (middle panel). The right panel displays backgating of CD7+ stem cells (black dots) into the CD45lowSSClowCD14–CD34+CD38- population (gray). (C) Absolute number of CD34+CD38– CD7+ cells according to case-control status. The dashed line denotes number of CD34+CD38– CD7+ cells required for aberrant CD7 expression. Five cells or more were regarded as a positive lower limit. Patients with <5 CD34+CD38– CD7+ cells are not shown as dots in the figure (case subjects, n = 20; control subjects, n = 33). The Pearson χ2 test was used to calculate P values, with the positive lower limit of 5 cells being the cutoff for yes/no for aberrant CD7 expression.
Flow cytometric analysis of aberrant CD7 expression within the stem cell subset. The CD34+CD38– stem cells were identified within the CD45lowSSClowCD14– cells (left panel), and the presence (A) or absence (B) of aberrant CD7+ cells were evaluated by using a bivariate plot of SSC-A vs CD7 (middle panel). The right panel displays backgating of CD7+ stem cells (black dots) into the CD45lowSSClowCD14–CD34+CD38- population (gray). (C) Absolute number of CD34+CD38– CD7+ cells according to case-control status. The dashed line denotes number of CD34+CD38– CD7+ cells required for aberrant CD7 expression. Five cells or more were regarded as a positive lower limit. Patients with <5 CD34+CD38– CD7+ cells are not shown as dots in the figure (case subjects, n = 20; control subjects, n = 33). The Pearson χ2 test was used to calculate P values, with the positive lower limit of 5 cells being the cutoff for yes/no for aberrant CD7 expression.
Combining NGS and FCM as a screening method for development of tMN
Given that both targeted NGS and aberrant expression of CD7 at the stem cell level are potentially efficacious screening methods to identify patients at high risk of developing tMN, we analyzed the performance of combining NGS and FCM as a screening tool. By using a simple binary outcome screening method, we would have been able to correctly identify 24 (66%) of 36 case subjects already at ASCT, whereas 8 (22%) of 36 control subjects would have been misclassified as cases. This suggests that NGS and FCM complement each other in a clinical setting, with the 2 methods combined in a simple algorithm yielding a sensitivity of 66% and a specificity of 78%. This corresponds to a positive predictive value of 75% and a negative predictive value of 70%.
Discussion
In this nested case-control study of patients developing tMN after ASCT, we found that the presence of CH before ASCT increases the risk of developing tMN with a factor of 5.9. The high number of tMN patients with available biobanked cells from leukapheresis products allowed for a robust estimate for development of tMN. Our data are in line with recently published data by Young et al35 who found a factor 5.4 increase in the risk of developing AML associated with detectable CH at a VAF of ≥0.01, as well as the ability to detect the presence of low VAF mutations several years before onset of tMN.36,37 To the best of our knowledge, the current study is the first to show that aberrant CD7 expression on the stem cell population from leukapheresis products is associated with an almost sevenfold increase in risk of developing tMN. Furthermore, we found that patients with tMN were poorer stem cell mobilizers during leukapheresis compared with control subjects. Collectively, these findings show that at ASCT, patients with low VAF mutations associated with myeloid neoplasms, or with aberrant CD7 expression within the stem cell subset, are at high risk of developing tMN.
Intriguingly, our results suggest that FCM is a highly accurate method for early identification of patients at risk of developing tMN, and we have shown that both NGS and FCM are useful methods in the early identification of patients at high risk of developing tMN. Importantly, these methods identified different patients in the tMN cohort, overlapping in just 5 patients, and only 3 control subjects displayed aberrant CD7 expression, all with a minimum of 10 years of follow-up (supplemental Table 1). All 5 patients with both detectable CH and expression of CD7 developed tMN, alluding to coexpression of CH and CD7 as a potential future predictor of tMN. However, the data require validation in a larger and independent cohort and cannot yet be considered for clinical evaluation of risk of tMN before ASCT. Cell sorting of CD7+ stem cells is needed for molecular and functional studies and is a prerequisite to unravel their potential role in the pathogenesis of tMN.
Four cases did not have their samples analyzed by FCM due to sparse sample material, suggesting that the actual predictive potential of our simple binary outcome screening could be even higher than reported here.
Immunophenotypic characterization of malignant stem cells has been studied in patients with AML and MDS, and the fraction of such aberrant marker–positive cells has been shown to hold prognostic impact at diagnosis and during treatment.38 Moreover, antigen targeting is an appealing approach in cancer treatment.29,39 Routinely, leukapheresis products are evaluated by FCM to enumerate CD34+ cells, and simultaneous evaluation of aberrant CD7 expression on CD38+CD34– cells would be feasible in most laboratories as performed in diagnostic and MRD settings of AML and MDS, as shown by Zeijlemaker et al.28
Targeted NGS offers the advantage of accurate prediction of which specific mutations confer an actual risk of tMN, albeit this perspective will be consolidated in the future. The accumulating evidence pointing to the increased risk of tMN in patients with CH who are treated with ASCT makes close monitoring with early intervention an alluring and feasible option. Third, results could imply reevaluation of the indication for ASCT for patients with tMN-associated mutations who were allocated for ASCT for an incurable neoplasm (eg, patients with multiple myeloma). However, for a considerable group of patients, the decision to pursue treatment with ASCT would be unaffected by the presence of CH in the leukapheresis product. Undoubtedly, future prognostic models and more accurate prediction tools will guide clinical decision-making.
One patient sequenced at ASCT and at tMN showed that CH, present at low VAF at ASCT, had the potential to expand toward tMN. This finding challenges the validity of the VAF 0.02 limit to qualify for the CHIP diagnosis, especially considering the fact that we have shown the relevance of mutations with VAFs as low as 0.003. Thus, our findings suggest both a biological and a clinical impact of low VAF mutations detected by using NGS. To further scrutinize the pathogenesis of progression from CH to tMN, the subject of future research should focus on the underlying mechanisms behind the evolutionary potential and interplay of individual mutations.
In the current study, we used a commercially available panel limited to 30 genes, as used in routine diagnostics of MDS and AML at our institution. Specifically, this panel did not include PPM1D, which in recent publications has been strongly correlated with tMN.40,41 Notably, in our cohort, case subjects could have had an undiagnosed myeloid malignancy that was not identified in histologic analysis of bone marrow biopsy specimens before ASCT, and the apparent development of tMN could thus represent progression of an undiagnosed preexisting condition. In particular, patient 1179 had a classic CALR mutation at a VAF of 0.40, strongly suggesting that this patient had an undiagnosed myeloproliferative neoplasm before treatment with ASCT; hence, it cannot be excluded that the tMPN diagnosis in fact represents progression from an undiagnosed myeloproliferative neoplasm. Furthermore, we did not have available samples to evaluate whether variants reported could represent potential germline mutations, in particular mutations present at VAFs of 0.44 to 0.55 and >0.95, and it is therefore possible that our results could contain rare germline variants.
Preferentially, data from NGS analyses on tMN samples would enable evaluation of the clonal evolution of mutations present at ASCT. Theoretically, the clones detected at ASCT might not undergo evolution themselves and cause tMN but may act in a more complex interplay with other molecular aberrations. Also, we expect that the incidence of tMN in our cohort of ASCT patients, in particular tMDS, could be underestimated. Mild persistent cytopenia, which can be an early sign of tMDS, in patients who have been treated with ASCT may to some extent be accepted without performing diagnostic analyses. Furthermore, patients with relapse of their primary disease could be spared from undergoing new biopsies, as curative intent could be out of reach. Finally, the diagnosis of tMDS could have been achieved in other ways, such as identified according to MDS-specific cytogenetic abnormalities.
In conclusion, we performed a nested case-control study on patients who developed tMN after ASCT. We found that screening for mutational status and aberrant expression of CD7 on hematopoietic stem cells at time of ASCT can identify patients at high risk of developing tMN. We further established that mutations were relevant, and predictive, of tMN at VAFs lower than 0.02. New research into the predictive potential of the individual mutations in the spectrum of CH, preferably combined with sequential NGS analyses to map the potential evolution of early leukemic clones over time, will drive future clinical decision-making.
For original data, please contact the corresponding author (Maja Ludvigsen; e-mail: majlud@rm.dk).
Acknowledgments
The authors thank Henriette Rugholm Petersen for expert technical assistance. They also thank Michael Roost Clausen and Signe Borgquist for providing clinical data.
This work was supported by grants from the Danish Cancer Society, the Wørzner Memorial Foundation for Cancer Research, and the Agriculturalist of ‘Ølufgård’-Peder Nielsen Kristensen’s Memorial Foundation.
Authorship
Contribution: J.F.S., A.S.R., and M.L. designed the study, interpreted the data, and wrote the manuscript; J.F.S., A.S.R., and P.H. identified patients for the study; G.B.K. revised the tMN biopsy samples; C.A.R., M.B., L.H.E., and I.K.L.E. designed and conducted the flow cytometric evaluations; A.A., M.C.H., and J.F.S. performed the NGS analyses as well as interpretation of VAF levels; and all authors critically read and approved the manuscript before publication.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Maja Ludvigsen, Department of Hematology, Aarhus University Hospital and Department of Clinical Medicine, Aarhus University, Palle Juul-Jensens Blvd 99, 8200 Aarhus N, Denmark; e-mail: majlud@rm.dk.