

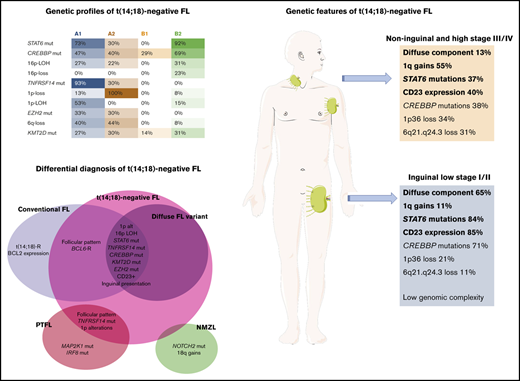
Key Points
t(14;18)− FL is genetically distinct from t(14;18)+ FL, NMZL, and PTFL.
STAT6 mutations are frequent and co-occur with TNFRSF14 and/or CREBBP alterations, revealing alternative oncogenic pathways.

Abstract
Fifty-five cases of t(14;18)− follicular lymphoma (FL) were genetically characterized by targeted sequencing and copy number (CN) arrays. t(14;18)− FL predominated in women (M/F 1:2); patients often presented during early clinical stages (71%), and had excellent prognoses. Overall, t(14;18)− FL displayed CN alterations (CNAs) and gene mutations carried by conventional t(14;18)+ FL (cFL), but with different frequencies. The most frequently mutated gene was STAT6 (57%) followed by CREBBP (49%), TNFRSF14 (39%), and KMT2D (27%). t(14;18)− FL showed significantly more STAT6 mutations and lacked MYD88, NOTCH2, MEF2B, and MAP2K1 mutations compared with cFL, nodal marginal zone lymphoma (NMZL), and pediatric-type FL (PTFL). We identified 2 molecular clusters. Cluster A was characterized by TNFRSF14 mutations/1p36 alterations (96%) and frequent mutations in epigenetic regulators, with recurrent loss of 6q21-24 sharing many features with cFL. Cluster B showed few genetic alterations; however, a subgroup with STAT6 mutations concurrent with CREBBP mutations/16p alterations without TNFRSF14 and EZH2 mutations was noted (65%). These 2 molecular clusters did not distinguish cases by inguinal localization, growth pattern, or presence of STAT6 mutations. BCL6 rearrangements were demonstrated in 10 of 45 (22%) cases and did not cluster together. Cases with predominantly inguinal presentation (20 of 50; 40%) had a higher frequency of diffuse growth pattern, STAT6 mutations, CD23 expression, and a lower number of CNAs, in comparison with noninguinal cases (5.1 vs 9.1 alterations per case; P < .05). STAT6 mutations showed a positive correlation with CD23 expression (P < .001). In summary, t(14;18)− FL is genetically a heterogeneous disorder with features that differ from cFL, NMZL, and PTFL.
Introduction
The genetic hallmark of conventional follicular lymphoma (cFL) is the chromosomal translocation t(14;18)(q32;q21) present in ∼85% to 90% of cases, resulting in the constitutive overexpression of the antiapoptotic protein BCL2.1 In recent years, the genetic landscape of cFL has been characterized comprehensively, revealing that germinal center (GC)–derived lymphomas carry frequent mutations in histone-modifying genes.2-5 The histone methyltransferases KMT2D and EZH2; the histone acetyltransferases CREBBP, EP300, and MEF2B; and the tumor necrosis factor receptor TNFRSF14 are the most commonly mutated genes. cFL is also characterized by recurrent gains in the chromosomal regions 1q, 2p15, 7, 8q, 12q, 18p, and 18q, as well as losses in 1p36, 3q, 6q, 9p, 10q, 11q, 13q, and 17p.6-8 Sites of recurrent copy number neutral–loss of heterozygosity (CNN-LOH) have been detected on 6p, 16p, 12q, 1p36, 10q, and 6q.9 Interestingly, CREBBP and TNFRSF14 are also affected by frequent deletions or CNN-LOH alterations that produce biallelic inactivation in a considerable number of cases (15% and 23%, respectively).2,5
FL t(14;18)− comprises 10% to 15% of all FL cases. Although BCL2 staining is a standard diagnostic procedure, it is not a perfect surrogate marker for the translocation, given that BCL2 protein is also expressed in a subset of t(14;18)− FL.6,10,11 Furthermore, apparent lack of BCL2 expression can be observed in some FL with t(14;18) translocation related to altering of antibody binding by mutations.12 Genetically, t(14;18)− FL is less well characterized, and its pathogenesis is largely unknown. A gene expression profiling study comparing t(14;18)− and t(14;18)+ FLs showed signatures that point to subtle differences in the developmental stages of the neoplastic B cells, the usage of divergent oncogenic pathways, and the composition of the microenvironment.6 Specifically, GCB-cell signatures were enriched in t(14;18)+ FL, whereas ABC-like and post-GCB signatures were found to be overexpressed in t(14;18)− FL. Notably, t(14;18)− FLs showed evidence of ongoing somatic hypermutation, a feature of GCB-cells. It was therefore suggested that a subset of t(14;18)− FL may show the phenotype of a late GCB cell that has not yet exited the GC stage of differentiation. This latter observation was supported by a specific micro-RNA expression profile.13
A subset of t(14;18)− FLs shows a rearrangement affecting chromosomal band 3q27, which results in the deregulation of BCL6 expression.14 BCL6 translocation has been reported in grade 3B FLs (44%), which are more closely biologically related to diffuse large B-cell lymphoma than to other FLs.15,16 Nevertheless, the frequency of t(3;14)(q27;q32) is also high (22%) in t(14;18)− FL grades 1 and 2,17,18 suggesting an important role for BCL6 in a subgroup of t(14;18)− FL.19
A group of t(14;18)− FL with a predominantly diffuse growth pattern and 1p36 chromosomal deletion was described by Katzenberger et al.20 These cases were characterized by expression of CD23 and frequent loss of CD10 expression. Clinically, inguinal presentation predominates with early clinical stages (stages I and II) at diagnosis. Recently, it has been shown that these cases have frequent mutations of STAT6, CREBBP, KMT2D, and TNFRSF14 genes; whether these cases represent a distinct subgroup within FL is as yet not clear.21,22
The diagnosis of t(14;18)− FL cases is difficult, and the differential diagnosis with reactive conditions, nodal marginal zone lymphomas (NMZLs)23,24 and pediatric-type FL (PTFL) is challenging.25-27 Because previous studies on t(14;18)− FL have included relatively few cases,21,22 most of which showed BCL2 expression, we sought in this study to characterize the genetic profile of a large cohort of t(14;18)− FL, mostly without BCL2 expression, to identify specific genetic characteristics and correlate them with morphology, immunophenotype, and clinical features.
Material and methods
Lymphoma samples and clinical data
Fifty-five cases of t(14;18)− nodal FL, grades 1 to 3A were collected from the University of Tübingen (Germany), National Cancer Institute, National Institutes of Health (Bethesda, MD), Hospital Clinic (Barcelona, Spain), Hospital Henri-Mondor (Creteil, France), Wilheminenspital (Vienna, Austria), University Hospital of Wales (Cardiff, United Kingdom), and University of Siena (Italy). The cases were reviewed by 5 of the authors (E.C., B.G.F., O.B., E.S.J., and L.Q.-M.), according to the criteria of the 2017 World Health Organization classification.1 Ten of the cases have been published.26 All cases had at least 50% tumor cells. The morphology, growth pattern, cytology, and immunohistochemical staining for CD20, CD79a, BCL6, BCL2, CD10, CD3, MIB1, and CD23 were evaluated in formalin-fixed, paraffin-embedded (FFPE) tissue sections. The study was conducted in accordance with the Declaration of Helsinki and was approved by the local Ethics Review Committee and the institutional review board panels of the contributing institutions (UKT 348/2020B0).
DNA extraction and clonality analysis
DNA was extracted from FFPE tissue after dewaxing and protein K digestion, applying either an FFPE DNA Tissue Kit (Qiagen, Valencia, CA) or the Maxwell FFPE Tissue LEV DNA Purification Kit (Promega, Madison, WI) according to the manufacturer’s protocol. Polymerase chain reaction amplifications for detecting monoclonal IGH/IGκ chain gene rearrangements were performed in selected cases according to the BIOMED-2 protocol (supplemental Methods).28
Fluorescence in situ hybridization
Interphase fluorescence in situ hybridization (FISH) analysis for the detection of BCL2 and BCL6 translocations was performed with LSI BCL2 or BCL6 Dual-Color Break-Apart Probes (Vysis-Abbott Molecular, Wiesbaden, Germany). In some cases, FISH for the t(14;18)/IGH/BCL2 translocation was performed as part of the diagnostic workup.
CN analysis
DNAs extracted from FFPE were hybridized on the MIP-assay using Oncoscan FFPE according to standard protocols (Thermo Fisher Scientific, Schwerte, Germany). For comparison, previously published CN data on 35 cases of NMZL24 and 42 of PTFL26 were used, as well as data on t(14;18)+ and t(14;18)− FL (for details see supplemental Methods).6,7 Gains and losses and CNN-LOH regions were evaluated and visually inspected by using Nexus Biodiscovery version 9.0 software (Biodiscovery, Hawthorne, CA). Chromothripsis-like patterns were evaluated according to previously described criteria.29 The human reference genome was GRCh37/hg19. The CNAs with a minimum size of 100 kb and CNN-LOHs larger than 5 Mb were considered informative.
NGS analysis
Next generation sequencing (NGS) analysis was performed on the Ion Torrent PGM and S5 from Thermo Fisher Scientific. NGS libraries were amplified by using 2 primer pools of 2 Ion AmpliSeq Custom Panels covering 20 genes that have been shown to be frequently mutated in FL (panels NGS1 and NGS2; supplemental Tables 1 and 2).2,5,30,31 The custom panels were designed with the Ion AmpliSeq Designer from Thermo Fisher Scientific (version 3.4). The library preparation, sequencing, and raw data analyses are described in the supplemental Methods.
Targeted resequencing and Sanger sequencing
For validation of the NGS results, variants with low allelic frequencies (<15%) were reanalyzed as single amplicons by using a targeted resequencing approach on the Ion Torrent PGM or S5. Variants with high allelic frequencies (≥15%) were verified with Sanger Sequencing.26 The primer design process is described in the supplemental Methods, and the primers are shown in supplemental Tables 3 and 4.
Statistical analysis
Differences in the distribution of individual parameters among subsets of patients were analyzed by Fisher’s exact test for categorized variables, and the Student t test for continuous variables. Nonparametric tests were applied when necessary. Details on the statistical and clustering analyses are provided in the supplemental Methods.
Results
Clinical and morphological findings
The clinical and morphological information is summarized in Table 1. A total of 55 patients were included in the study, of which 18 were men and 37 were women, representing an M/F ratio of 1:2, with a median age of 61 years (range, 33-87 years). Inguinal lymph nodes (LNs) were most frequently involved (30 of 55, 55%; isolated 20 of 50 cases, 40%), followed by axillary LNs (13 of 55; 24%), cervical LNs (8 of 55; 15%), and others (4 of 55; 7%). Of the 43 patients with available clinical information, 29 had low-stage disease at presentation (I-II; 71%); whereas 12 patients had high-stage disease (III-IV; 29%). Thirty-seven patients (86%) are alive without evidence of disease with a median follow-up of 53 months (range, 3-242 months). Nine patients showed 1 or more relapses, of which 7 are alive with no evidence of disease. Only 3 of 43 (7%) patients died of disease, 1 with transformation to diffuse large B-cell lymphoma.
Morphologically, a follicular growth pattern was observed in 33 of 55 cases (60%), a follicular and diffuse pattern in 8 of 55 cases (15%), and a predominantly diffuse pattern in 14 of 55 cases (25%) (Figure 1). Forty-eight cases (87%) were considered to have grade 1 and 2 cytology, and 7 cases (13%) were considered to have grade 3A cytology. Immunohistochemically, all cases were CD20+ and/or CD79a+. The GC markers BCL6 and/or CD10 were expressed in most cases (Figure 1C,J-K). CD10 was negative in 9 cases (9 of 55; 16%). BCL2 was negative in 50 cases using either the 100/D5 clone (Figure 1B,H) or the alternative clone E17 (Figure 1F,I). Five cases showed BCL2 staining. CD23 was expressed in the tumor cells in 33 of 55 cases (59%; Figure 1D,L-M). The proliferation rate showed a broad range with a median proliferation of 20% in grades 1 and 2 and 65% in grade 3A (Figure 1E).
![Histologic features of t(14;18)−FL. (A-F) Case FL7, group B2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E]; original magnification, ×50). (B) The follicles are BCL2− with the 100/D5 clone (immunoperoxidase; original magnification, ×50). (C) The follicles are CD20+. (D) The tumor cells are CD23+. (E) MIB1 stain demonstrates low proliferation within the follicles without polarization. (F) The tumor cells are also negative with the alternative BCL2 antibody clone E17 (C-F; immunoperoxidase; original magnification, ×100). (G-M) Case FL11, group A1. (G) Inguinal LN with atypical lymphoid proliferation with follicular (right side) and diffuse (left side) growth pattern (H&E; original magnification, ×25). (H-I) The cells are negative with the BCL2 100/D5 clone and the E17 clone. (J-K) The tumor cells are CD10+ in the follicular and diffuse areas. (L-M) CD23 is strongly expressed in the follicular and diffuse areas (H-M; immunoperoxidase; original magnification, ×100).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/22/10.1182_bloodadvances.2020002944/2/m_advancesadv2020002944f1.png?Expires=1770096918&Signature=nNt7gOmiTNny4ACUt~zxf5M~utO180dp0n1~rZ8kjbTcbpFYdQahGj-BrBRwUY1Feo9dEaSWXl3KQAFnTkNrat~BZkxkA2ReYW5NnvDHKL4Ak8TOljw4nx8gnwMRJiHUTk628a3mCYUeHTP00kcgaaezzi577ByRSDjzwdO-ccITQBBHtiAxCRNOVpB544v1OlaDnT8ptsn5C32CNOiiwt1IGK4d~OT9XkHmZlsBOpzxVJ8cLtjM0mqMGvnADtDEjdAT72JN5fpTu2O7I5-M~iQx6okCgjXaPoqJ~un8a0OLRlJD9Rl3Q4Q~2XJNub4BRoZColxjZLlovzIPsGDh7A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Histologic features of t(14;18)−FL. (A-F) Case FL7, group B2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E]; original magnification, ×50). (B) The follicles are BCL2− with the 100/D5 clone (immunoperoxidase; original magnification, ×50). (C) The follicles are CD20+. (D) The tumor cells are CD23+. (E) MIB1 stain demonstrates low proliferation within the follicles without polarization. (F) The tumor cells are also negative with the alternative BCL2 antibody clone E17 (C-F; immunoperoxidase; original magnification, ×100). (G-M) Case FL11, group A1. (G) Inguinal LN with atypical lymphoid proliferation with follicular (right side) and diffuse (left side) growth pattern (H&E; original magnification, ×25). (H-I) The cells are negative with the BCL2 100/D5 clone and the E17 clone. (J-K) The tumor cells are CD10+ in the follicular and diffuse areas. (L-M) CD23 is strongly expressed in the follicular and diffuse areas (H-M; immunoperoxidase; original magnification, ×100).
Histologic features of t(14;18)−FL. (A-F) Case FL7, group B2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E]; original magnification, ×50). (B) The follicles are BCL2− with the 100/D5 clone (immunoperoxidase; original magnification, ×50). (C) The follicles are CD20+. (D) The tumor cells are CD23+. (E) MIB1 stain demonstrates low proliferation within the follicles without polarization. (F) The tumor cells are also negative with the alternative BCL2 antibody clone E17 (C-F; immunoperoxidase; original magnification, ×100). (G-M) Case FL11, group A1. (G) Inguinal LN with atypical lymphoid proliferation with follicular (right side) and diffuse (left side) growth pattern (H&E; original magnification, ×25). (H-I) The cells are negative with the BCL2 100/D5 clone and the E17 clone. (J-K) The tumor cells are CD10+ in the follicular and diffuse areas. (L-M) CD23 is strongly expressed in the follicular and diffuse areas (H-M; immunoperoxidase; original magnification, ×100).
IGH/IGκ clonality and FISH analysis
All 26 cases analyzed were monoclonal. FISH analysis demonstrated the absence of BCL2 rearrangement in all 55 cases, using the BCL2 break-apart probe (42 cases) and/or t(14;18) dual-color, dual-fusion probes (24 cases). BCL6 rearrangements were identified in 10 of 45 (22%) cases.
CN and CNN-LOH alterations in t(14;18)− FL
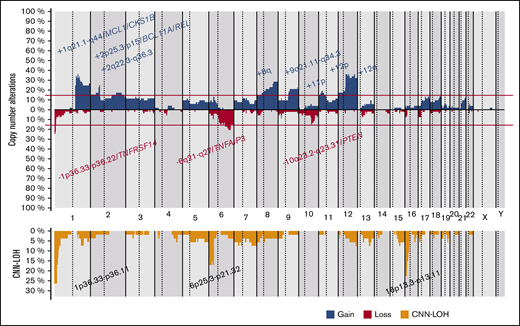
CN-analysis of t(14;18)− FL identified 380 CNAs in 52 of 53 cases, with a mean of 7.2 imbalances per case (range, 0-28; Figure 2; supplemental Table 5). Specifically, 200 gains, 152 losses, 24 amplifications, and 4 homozygous deletions were detected. The most recurrently gained regions (≥15% of the cases) were identified at 1q21.1-q44, 2p25.3-p15/REL&BCL11A, 2q22.3-q36.3, 8q, 9q21.11-q34.3, 11p, 12p, and 12q. Recurrent regions of loss (≥15% of the cases) were observed at 1p36.33-p36.22/TNFRSF14, 6q21-q27/TNFAIP3, and 10q23.2-q23.31/PTEN. Recurrent amplifications occurred at 1q21.2-q25.3 targeting MCL1, CKS1B, and IL6R genes (2 cases) and 2p16.1-p15 targeting BCL11A/REL (2 cases). Interestingly, focal 1p36.32 homozygous deletions, including TNFRSF14 were observed in 2 cases, whereas a single case had the 9p21.3 homozygous deletion including the CDKN2A/B locus. Chromothripsis-like patterns were observed in 4 cases, targeting chromosomes 1 (2 cases), 3, and 10. One hundred twenty regions of CNN-LOH were also detected, with 1p36.33-p36.11/TNFRSF14 (26%), 16p13.3-p12.32/CREBBP (23%), and 6p25.3-p21.32 (15%) being the most affected regions.
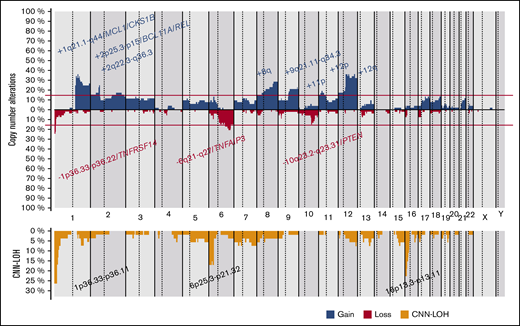
Copy number profile of 53 t(14;18)−FLs. Global copy number (top panel) and copy number neutral loss of heterozygosity (CNN-LOH; bottom panel) profile. The x-axis depicts chromosome positions with dotted lines pointing out centromeres. The y-axis indicates frequency of the genomic aberration among the analyzed cases. Each probe is aligned with chromosomes from 1 to Y and p to q. Gains are depicted in blue and losses in red, and regions of CNN-LOH are represented in yellow. Recurrent CN and CNN-LOH regions (>15% of cases) are indicated.
Copy number profile of 53 t(14;18)−FLs. Global copy number (top panel) and copy number neutral loss of heterozygosity (CNN-LOH; bottom panel) profile. The x-axis depicts chromosome positions with dotted lines pointing out centromeres. The y-axis indicates frequency of the genomic aberration among the analyzed cases. Each probe is aligned with chromosomes from 1 to Y and p to q. Gains are depicted in blue and losses in red, and regions of CNN-LOH are represented in yellow. Recurrent CN and CNN-LOH regions (>15% of cases) are indicated.
Identification of recurrent mutations by targeted NGS
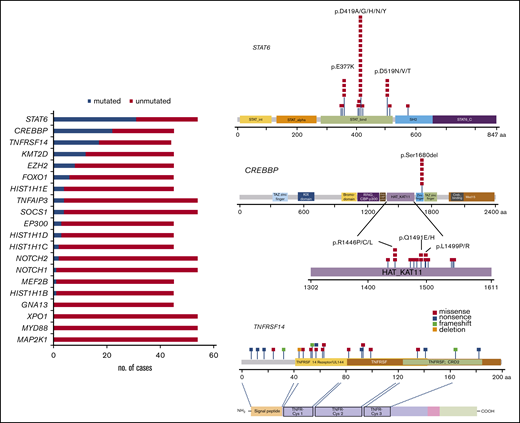
All cases were analyzed by NGS. The analyses of 45 cases were informative with both gene panels, whereas 9 with low DNA quality (<200 bp) were informative only with panel 2. One case was not informative (FL23). The mean average read depth of the NGS sequence analysis was 3 551 (range, 114-26 723). In the 54 evaluable cases, 133 mutations were identified (supplemental Table 6) with a mean of 2.3 mutations per case. The most frequently mutated gene was STAT6, with 33 mutations in 31 patients (31 of 54; 57%) with a median variant allele frequency (VAF) of 19% (range, 4%-58%), followed by 24 CREBBP mutations in 22 patients (49%) with a median VAF of 25% (range, 3%-60%), 21 TNFRSF14 mutations in 17 patients (39%) with a median VAF of 17% (range, 5%-71%), and KMT2D mutations in 12 patients (27%) with a median VAF of 23% (7%-44%; Figure 3; left). Additional mutated genes were EZH2 (8 of 45; 18%), FOXO1 (6 of 45; 13%), HIST1H1E (4 of 45; 9%), TNFAIP3 (4 of 54; 7%), EP300 (3 of 45; 7%), HIST1H1D (3 of 45; 7%), SOCS1 (4 of 54; 7%), HIST1H1C (2 of 45; 4%), NOTCH2 (2 of 54; 4%), NOTCH1 (1 of 54; 2%), MEF2B (1 of 45; 2%), and HIST1H1B (1 of 45; 1%). No mutations were identified in GNA13, XPO1, MYD88, and MAP2K1.
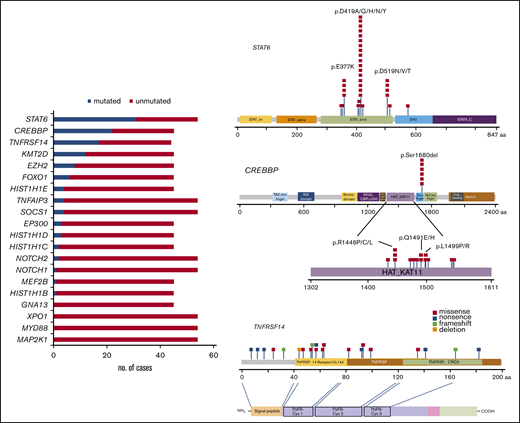
Mutational landscape of 54 t(14;18)−FLs. Bar graphs (left) show the 20 genes analyzed, and the results are given in number of mutated (blue) and nonmutated (red) cases. A diagram of the relative positions of driver mutations (right) is shown for STAT6, CREBBP, and TNFRSF14. The x-axis indicates the amino acid positions. The approximate location of somatic mutations identified in each gene is indicated. STAT6 mutations are mainly in the DNA binding domain with 3 hot spots. Mutations in CREBBP are mainly identified in the HAT-KAT domain and a hot spot in p.S1680del. Mutations in TNFRSF14 are identified mainly in the 3 first exons. The protein with its different domains (bottom) and the cysteine repeats TNFR-Cys 1-3. Domains of the protein are represented according to the Uniprot database (www.uniprot.org).
Mutational landscape of 54 t(14;18)−FLs. Bar graphs (left) show the 20 genes analyzed, and the results are given in number of mutated (blue) and nonmutated (red) cases. A diagram of the relative positions of driver mutations (right) is shown for STAT6, CREBBP, and TNFRSF14. The x-axis indicates the amino acid positions. The approximate location of somatic mutations identified in each gene is indicated. STAT6 mutations are mainly in the DNA binding domain with 3 hot spots. Mutations in CREBBP are mainly identified in the HAT-KAT domain and a hot spot in p.S1680del. Mutations in TNFRSF14 are identified mainly in the 3 first exons. The protein with its different domains (bottom) and the cysteine repeats TNFR-Cys 1-3. Domains of the protein are represented according to the Uniprot database (www.uniprot.org).
STAT6 gene mutations were nonsynonymous and targeted the STAT6 DNA binding domain except for 1 mutation (p.R578Q) located in the SH2 domain (Figure 3; right). The most common recurrent mutation was the hot spot in amino acid residue 419 (p.D419A/G/H/N/Y) found in 17 cases (52%) followed by 4 cases (12%) each in amino acid residues 377 and 519 (p.E377K and p.D519N/V/T). CREBBP missense mutations were mostly identified in the HAT-KAT domain (18 mutations; 75%), whereas 6 mutations (25%) were identified in a hot spot in the zinc finger domain, causing the loss of S1680 in a 3-bp in-frame deletion, suggesting an important functional role for this uncharacterized serine. TNFRSF14 mutations included 10 missense mutations, 7 nonsense mutations, 1 deletion (deletion of 3 nucleotides), and 3 frameshift mutations (insertion of 1 or 2 nucleotides). Forty-four cases (44 of 54; 81%) displayed at least 1 mutation, and 5 cases (5 of 45; 11%) showed no mutations. Sixty-two selected mutations were further verified, by either Sanger sequencing or targeted resequencing. Analysis of SIFT and Polypen 2 predicted a damaging effect in 130 of the 133 mutations (supplemental Table 6).
Three genes were frequently biallelically inactivated because of concomitant loss/CNN-LOH and mutations. Of 17 cases with TNFRSF14 mutations, 12 were affected by local 1p36 aberrations (5 losses and 7 CNN-LOH). Of 22 cases with CREBBP mutations, 8 had concomitant CNN-LOH of the 16p region and 1 had a 16p loss. Finally, all 4 cases with TNFAIP3 mutations had the 6q alteration (3 with losses and 1 with CNN-LOH).
Genetic comparison of t(14;18)− FL with cFL, NMZL, PTFL, and DTFL
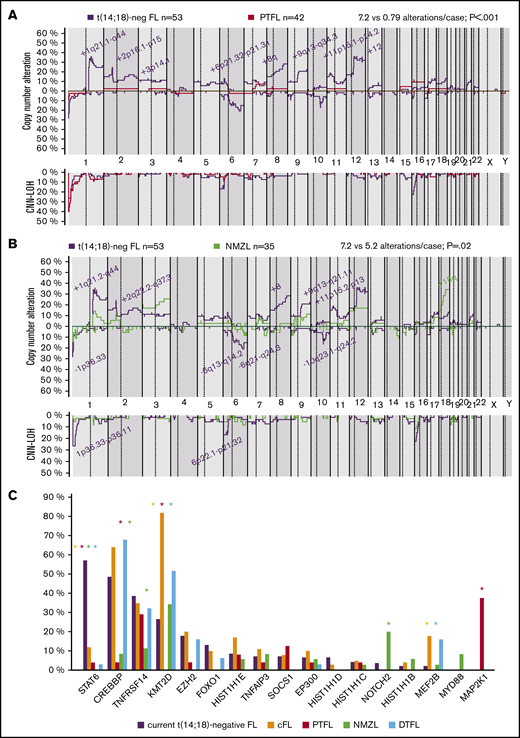
The t(14;18)− FL CN profile was compared with previously published t(14;18)+ and t(14;18)− FL, PTFL, and NMZL profiles.6,7,24,26 The t(14;18)− FL in this series had more 1q21.1-q44 and 8q gains in comparison with t(14;18)+ FL and more gains of 8q in comparison with other t(14;18)− cases, independent of BCL2 expression (supplemental Figure 1). In terms of CNN-LOH, only 1p36 regions were more frequently altered in t(14;18)− FL in comparison with t(14;18)+ FL (26% vs 8%; P = .004). Compared with PTFL, t(14;18)− FL was more complex in terms of CNA (7.2 vs 0.79 alterations per case; P < .01; Figure 4A). t(14;18)− FL had higher genetic complexity than NMZL (7.2 vs 5.2 alterations per case; P = .02), but no significant differences were found. Nevertheless, gains of 18q were more frequently observed in cases of NMZL (Figure 4B).
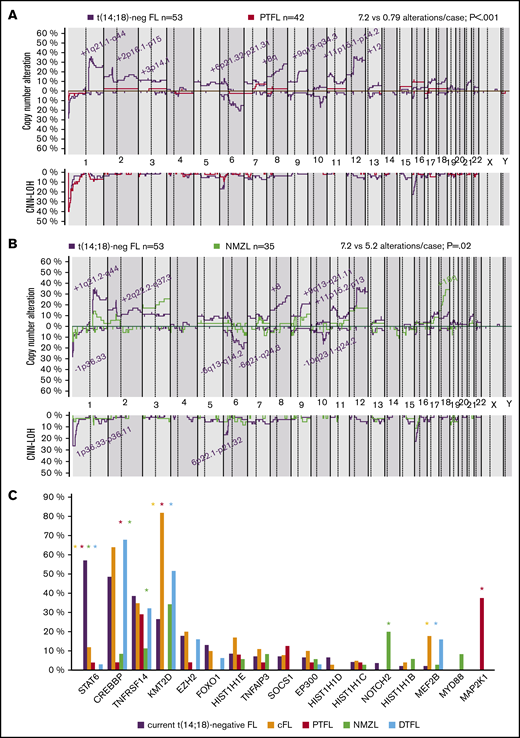
Genetic comparison of t(14;18)−FL cases with cFL, PTFL, NMZL, and DTFL. (A) CN comparison of t(14;18)− FL vs PTFL.26 Comparative plot of CN and CNN-LOH between 53 t(14;18)− FL cases and 42 PTFL cases. Significantly different regions are indicated in the plot, and the color denotes the enriched group (Fisher’s exact test; adjusted P < .05; minimum of 3 cases). (B) CN comparison of t(14;18)− FL vs NMZL. Comparative plot of CN and CNN-LOH between 53 cases of t(14;18)− FL and 35 of NMZL. No significant differences were observed between groups (Fisher’s exact test; adjusted P < .05; minimum of 3 cases). Nonetheless, some differences could be detected using raw nonadjusted P values. These differentially affected regions are indicated in the plot, and the color denotes the enriched group (Fisher's exact test; P < .05; minimum of 3 cases).24 (C) Comparison of recurrent mutations of t(14;18)− FL vs cFL, PTFL, NMZL, and DTFL. Frequencies of recurrently mutated genes (>5% cases) in 45 t(14;18)− cases and 22 cFL,5 24 PTFL,25 35 NMZL,24 and 31 DTFL.32 Asterisk color indicates significant differences between t(14;18)− FL and the associated color group (Fisher’s exact test; P < .05).
Genetic comparison of t(14;18)−FL cases with cFL, PTFL, NMZL, and DTFL. (A) CN comparison of t(14;18)− FL vs PTFL.26 Comparative plot of CN and CNN-LOH between 53 t(14;18)− FL cases and 42 PTFL cases. Significantly different regions are indicated in the plot, and the color denotes the enriched group (Fisher’s exact test; adjusted P < .05; minimum of 3 cases). (B) CN comparison of t(14;18)− FL vs NMZL. Comparative plot of CN and CNN-LOH between 53 cases of t(14;18)− FL and 35 of NMZL. No significant differences were observed between groups (Fisher’s exact test; adjusted P < .05; minimum of 3 cases). Nonetheless, some differences could be detected using raw nonadjusted P values. These differentially affected regions are indicated in the plot, and the color denotes the enriched group (Fisher's exact test; P < .05; minimum of 3 cases).24 (C) Comparison of recurrent mutations of t(14;18)− FL vs cFL, PTFL, NMZL, and DTFL. Frequencies of recurrently mutated genes (>5% cases) in 45 t(14;18)− cases and 22 cFL,5 24 PTFL,25 35 NMZL,24 and 31 DTFL.32 Asterisk color indicates significant differences between t(14;18)− FL and the associated color group (Fisher’s exact test; P < .05).
Recurrent mutations (>5% cases) in t(14;18)− FL identified in this study were compared to previously published NGS data for t(14;18)+ FL, PTFL, NMZL, and duodenal-type FL (DTFL).5,24,25,32 This comparison showed significant differences among these entities (Figure 4C). t(14;18)− FL showed significantly more STAT6 mutations, whereas t(14;18)+ FL and DTFL had more KMT2D and MEF2B mutations. t(14;18)− FL lacked the MAP2K1 mutations typical of PTFL, and the NOTCH2 mutations characteristic of NMZL, whereas PTFL and NMZL had practically no STAT6 and CREBBP mutations.
Genetic subgroups of t(14;18)− FL
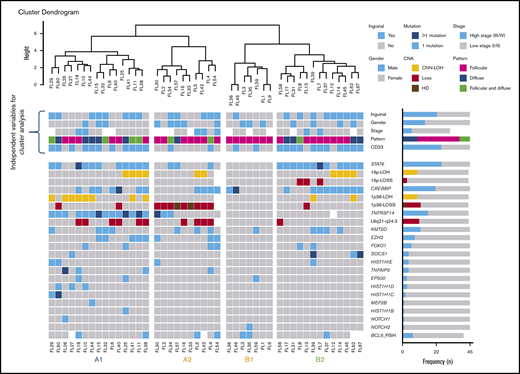
Unsupervised cluster analysis was performed in 45 cases in which mutational analysis of all genes investigated and CNA information were available (Figure 5). Correlation with independent clinicopathological features was also performed. The unsupervised analysis recognized 2 robust molecular clusters (supplemental Figure 2). Cluster A was characterized by TNFRSF14 mutations/1p36 alterations and mutations in other epigenetic regulators resembling cFL. In contrast, cluster B showed few genetic alterations. These 2 clusters did not distinguish cases according to inguinal localization, growth pattern, or presence of STAT6 mutations; nevertheless, within the clusters, 4 genetic profiles were identified and designated as A1, A2, B1, and B2.
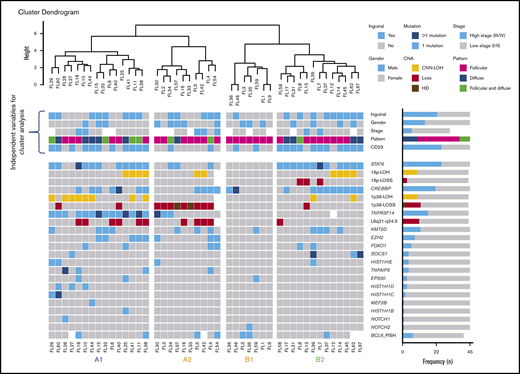
Cluster analysis of 45 t(14;18)−FL cases. Only patients with complete mutational analyses were included. Independent clinical and morphological variables for cluster analysis are depicted. The cluster analysis includes NGS results and frequent CNA affecting frequently mutated gene loci. Each column represents one patient and each line one specific analysis. On the right side of the figure, the frequency of the particular result of the analysis is shown. Four identified molecular groups are labeled as A1, A2, B1, and B2.
Cluster analysis of 45 t(14;18)−FL cases. Only patients with complete mutational analyses were included. Independent clinical and morphological variables for cluster analysis are depicted. The cluster analysis includes NGS results and frequent CNA affecting frequently mutated gene loci. Each column represents one patient and each line one specific analysis. On the right side of the figure, the frequency of the particular result of the analysis is shown. Four identified molecular groups are labeled as A1, A2, B1, and B2.
Group A1 included 15 cases and was characterized by the presence of the TNFRSF14 mutation/1p36 CNN-LOH (100%) and frequent STAT6 mutations (73%). Mutations in epigenetic regulator genes similar to cFL and deletions of 6q21 were mainly seen in this group. There was a female predilection (80%), with inguinal presentation in about half of the patients (53%), diffuse growth component (60%), and CD23 expression (73%). Group A2 (10 patients) was characterized by the presence of 1p36 losses (90%) with few STAT6 mutations (30%). Inguinal presentation was seen in 50% of the patients and follicular growth pattern predominated (70%). Group B2 included 13 patients who carried STAT6 mutations (92%) associated with CREBBP mutation/16p alterations (85%). These cases lacked TNFRSF14 and EZH2 mutations and showed few mutations in other epigenetic regulator genes, or 1p CNN-LOH and 6q deletions typical of cFL. Most patients were women presenting with disease in the inguinal region (69%), with follicular growth pattern (62%), and most often with CD23 expression (85%). Finally, group B1 (7 patients) was characterized by few genetic alterations, mainly noninguinal presentation with follicular growth pattern, lack of both STAT6 mutations and CD23 expression (P < .05), and no sex predilection. There was a positive correlation between STAT6 mutations and CD23 expression (P = .001).
Genetic features of patients with inguinal presentation
To further investigate the features of t(14;18)− FL patients with inguinal presentation, a comparative analysis was performed between patients with predominantly inguinal presentation (stage I; n = 18; stage II; n = 2) and those without inguinal presentation or inguinal stages III and IV (n = 30) (supplemental Figure 3). Patients with predominantly inguinal presentation had higher frequencies of diffuse growth component (65%), STAT6 mutations (84%), CD23 expression (85%), and mutations in epigenetic and chromatin modifier genes (94%) and a lower number of CNAs than did patients with noninguinal or inguinal stages III and IV (5.1 vs 9.1 alterations per case); and lack of 1q gains (P < .05). These results were also observed when a correlation analysis of the most recurrent molecular findings (>10% of mutations and >20% of CNAs) was performed in 55 cases (supplemental Figure 4).
BCL6 rearranged t(14;18)− FL
Ten cases carried BCL6 translocations with slight female predominance (male, 4; female, 6) and a median age at presentation of 64 years (range, 45-80 years). Five of 9 patients (56%) presented with advanced stage disease, in contrast to only 7 patients (24%) in the group without BCL6 translocation (P = .03). All cases showed a follicular growth pattern (Figure 6): 7 were grade 1 and 2, and 3 were grade 3A. Comparison between cases with and without the BCL6 translocation showed no significant differences in genetic complexity based on the number of CNAs (9.7 vs 7.1 alterations per case; P = .65). Cases with BCL6 rearrangement more frequently carried gains of 17q than did those without. Interestingly, alterations in 17p harboring the TP53 gene were found in 4 cases with BCL6 rearrangement (3 with losses and 1 with CNN-LOH) but only 1 of them carried a TP53 mutation (FL22). In contrast, only 1 case without BCL6 rearrangement had 17p loss, without mutation. The mutational landscape was otherwise similar in cases with and without BCL6 translocation. BCL6-rearranged cases did not cluster together.
![Morphological features and CN comparison of t(14;18)−FL BCL6-break positive vs BCL6-break negative cases. (A-H) Case FL5, group A2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E] stain; original magnification, ×50). (B) Higher magnification demonstrates that the infiltrate is mainly composed of centrocytes with a few scattered centroblasts (H&E stain; original magnification, ×400). (C) The tumor cells are CD20+ (immunoperoxidase, original magnification, ×50). (D) BCL2 is negative in the tumor cells but positive in the residual mantle zones and reactive T cells. (E) The tumor cells are BCL6+. FISH analysis using a BCL6 break-apart assay (inset) shows a signal constellation of 1 colocalized signal (yellow arrow) and 1 split signal (red and green arrows) consistent with gene rearrangement. Tumor cells are CD10+ (F), whereas the residual follicular dendritic cells are CD23+ (G). Note that the tumor cells are CD23−. (H) MIB1 shows a low proliferation rate without polarization in the germinal centers (D-H; immunoperoxidase; original magnification, ×100). (I) Comparative plot of CN and CNN-LOH between 9 BCL6 break positive cases and 35 BCL6 break negative cases. The only significantly different region is indicated in the plot, and the color denotes the enriched group (Fisher’s exact test; adjusted P < .05; minimum 3 cases).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/22/10.1182_bloodadvances.2020002944/2/m_advancesadv2020002944f6.png?Expires=1770096918&Signature=D2-rtRGTj2HwVG~BbU1qPwEg7zzVKFxqe6zirSuxMvSnbGAzqyRMxpXqWmrnfLKPbZ30~BZX4ETIBRWbeKTIi4APCrp3WC4i0aKhsmkVaM6JL5YbnnmNoNUmotMFqncYMjy0pA1-xfAyumHEJwjQEe50ORPIVfJ-Bu9dedXQdmzT7Ck0~Yj6UD9ZNr6~N6g6EVPdmHhY~BQYNKznx6WD1YeYkmKNp51nut2bpH7zdYbFH5mxKqP7as94y6ivZNoP2OPirrjSjEHvbif4yTANpOc23qZwVBjG345V5yriPvhTSVsd-aEYSeds7jlplqa6pwuMNqWWrNH8HAu5Xg~~BQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Morphological features and CN comparison of t(14;18)−FL BCL6-break positive vs BCL6-break negative cases. (A-H) Case FL5, group A2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E] stain; original magnification, ×50). (B) Higher magnification demonstrates that the infiltrate is mainly composed of centrocytes with a few scattered centroblasts (H&E stain; original magnification, ×400). (C) The tumor cells are CD20+ (immunoperoxidase, original magnification, ×50). (D) BCL2 is negative in the tumor cells but positive in the residual mantle zones and reactive T cells. (E) The tumor cells are BCL6+. FISH analysis using a BCL6 break-apart assay (inset) shows a signal constellation of 1 colocalized signal (yellow arrow) and 1 split signal (red and green arrows) consistent with gene rearrangement. Tumor cells are CD10+ (F), whereas the residual follicular dendritic cells are CD23+ (G). Note that the tumor cells are CD23−. (H) MIB1 shows a low proliferation rate without polarization in the germinal centers (D-H; immunoperoxidase; original magnification, ×100). (I) Comparative plot of CN and CNN-LOH between 9 BCL6 break positive cases and 35 BCL6 break negative cases. The only significantly different region is indicated in the plot, and the color denotes the enriched group (Fisher’s exact test; adjusted P < .05; minimum 3 cases).
Morphological features and CN comparison of t(14;18)−FL BCL6-break positive vs BCL6-break negative cases. (A-H) Case FL5, group A2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E] stain; original magnification, ×50). (B) Higher magnification demonstrates that the infiltrate is mainly composed of centrocytes with a few scattered centroblasts (H&E stain; original magnification, ×400). (C) The tumor cells are CD20+ (immunoperoxidase, original magnification, ×50). (D) BCL2 is negative in the tumor cells but positive in the residual mantle zones and reactive T cells. (E) The tumor cells are BCL6+. FISH analysis using a BCL6 break-apart assay (inset) shows a signal constellation of 1 colocalized signal (yellow arrow) and 1 split signal (red and green arrows) consistent with gene rearrangement. Tumor cells are CD10+ (F), whereas the residual follicular dendritic cells are CD23+ (G). Note that the tumor cells are CD23−. (H) MIB1 shows a low proliferation rate without polarization in the germinal centers (D-H; immunoperoxidase; original magnification, ×100). (I) Comparative plot of CN and CNN-LOH between 9 BCL6 break positive cases and 35 BCL6 break negative cases. The only significantly different region is indicated in the plot, and the color denotes the enriched group (Fisher’s exact test; adjusted P < .05; minimum 3 cases).
Discussion
In this study, we performed a genetic analysis of the largest series of t(14;18)− FL reported to date, with previous studies limited by a low number of cases6,21 or focused on cases with BCL2 expression11 or particular morphological features.20-22,33 We found that t(14;18)− FL is genetically a heterogeneous disease that is most common in women, usually presents in early clinical stages (stages I/II) at diagnosis, and has an excellent prognosis. Morphologically, cases with a pure follicular growth pattern predominated (60%), although a diffuse component was frequently observed (40%). We identified 2 molecular clusters with 4 genetic profiles that revealed key pathogenic mechanisms in t(14;18)− FL.
A rather characteristic finding of t(14;18)− FL was the high incidence of STAT6 mutations when compared with cFL (57% vs 11%-12%; P = .05).5,34 Although groups A1 and B2 shared the presence of STAT6 mutations (23 of 28 cases; 82%), each group had unique genetic features that suggested different pathogenic mechanisms. Group A1 was the closest to cFL and included mainly cases with TNFRSF14 mutations35 associated with 1p36 CNN-LOH or deletions (71%), supporting its role as a tumor-suppressor gene.26 In contrast to cFL, this group showed a significantly higher number of STAT6 mutations (73% vs 11%-12%; P < .05) and 1p36 CNN-LOH (53% vs 8%-15%; P < .05). Interestingly, 1p36 deletions defined group A2; however, no correlation with STAT6 mutations, inguinal presentation, or diffuse growth pattern was observed. Group B2, characterized by the co-occurrence of STAT6 with CREBBP mutations, in the absence of TNFRSF14 and EZH2 mutations suggests an alternative mechanism of epigenetic “addiction.” The inactivation of CREBBP alone facilitates oncogenic transformation by creating a permissive epigenetic environment in the GC that impairs the acetylation of the nonhistone proteins p53 and BCL6,36,37 whereas activated STAT6 contributes additional survival signals. Accordingly, the co-occurrence of STAT6 and CREBBP mutations has been noticed in BCL2− and BCL2+ FL,22,34 highlighting the cooperation of genes involved in GC fate and activated JAK-STAT signaling in the pathogenesis of some FL cases.5,36 Nevertheless, the results of the clustering analysis must be taken cautiously, because this is a relatively small series with limited molecular information that should be confirmed in further studies.
The cases in group B1 with few genetic alterations differed from the previous groups and raised the differential diagnosis of reactive conditions or NMZL.23,24 Nevertheless, these cases were all monoclonal: 2 cases each carried CREBBP mutations or BCL6 translocations and all expressed 1 or more GC markers. Moreover, NOTCH2 and MYD88 mutations described in 20% and 8% to 10%, respectively in NMZL, and gains of 18q were not detected in this cohort.24,38 Notably, cases in the B1 group as well as the other groups lack MAP2K1 mutations characteristically found in PTFL.25,31
Another interesting observation in this study was the positive correlation between STAT6 mutations and CD23 expression (P = .001). CD23 expression is rare in cFL but has been described as a characteristic feature of t(14;18)− FL.20,21 In normal B cells the most potent inducer of CD23 is interleukin-4 (IL-4) via STAT6 activation.39,40 In contrast, in STAT6 deficient B lymphocytes, IL-4 does not induce CD23 expression.40 Accordingly, our data indicate that the expression of CD23 may be secondary to activating STAT6 mutations, found to induce an exaggerated transcription of IL-4–responsive genes.34 Importantly, the expression of CD23 in this study predicted in most cases the presence of STAT6 mutation. Only 4 of 33 cases with CD23 expression lack demonstrable STAT6 mutations. Interestingly, 1 of these cases (FL26) carried a SOCS1 mutation, which is known to be upstream of STAT6 and contributes to constitutive STAT6 activation.41-43 Therefore, the expression of CD23 may serve as a diagnostic feature that should prompt assessment of the presence of STAT6 mutation and favors FL in the absence of t(14;18) chromosomal translocation. CD23 is usually negative in NMZL44-46 ; however, it has been reported in up to 28% of the cases in 1 study.47 In the light of these new findings, reevaluation of previous studies of CD23+ NMZL cases is warranted.
An important question is whether t(14;18)− FL with a diffuse pattern is a distinct entity. Our results do not support this contention and demonstrate that similar genetic and clinical features can be found in t(14;18)− FL cases with diffuse and/or follicular growth patterns, with or without inguinal presentation. An interesting finding, however, was the lower number of CNAs (P < .05) in the inguinal group, which correlates well with the usually localized presentation and indolent nature of the disease. The differences observed may be attributable to the inclusion criteria used in earlier studies, which were based on t(14;18)− FL cases with diffuse patterns, either alone20,33 or with the expression of CD2321 or the presence of 1p36 deletion.22 In the present study, we did not preselect the cases according to morphological characteristics and included all cases with a lack of t(14;18) chromosomal alteration confirmed by FISH analysis and mostly BCL2 negativity. Another difference was the low frequency of 1p36 deletions (28%) compared with previous studies.20,22 This discrepancy may be explained, in part, by the various sensitivities of the technologies used in the different studies: FISH vs CN analysis.
As reported previously,17,18 we confirmed the important role of BCL6 in a subgroup of patients with t(14;18)− FL (22%). Patients with BCL6 translocations, presented at more advanced clinical stages (56% vs 24%; P = .03) and accordingly showed a more complex genetic profile. Interestingly, in contrast to TNFRSF14 mutated cases, BCL6 transclocated, and CREBBP mutated cases were associated with a follicular growth pattern (100% and 68%, respectively), 2 master transcription regulators involved in the fate of the GC, suggesting that genetic alterations may influence the morphology of t(14;18)− FL.
Overall, t(14;18)− FL displays a typical cFL CNA profile, including aberrations of 1p/TNFRSF14, gains of 1q, gains of 2p16/REL/BCL11A, losses of 6q21-q24.3/SGK1/TNFAIP3, gains of 8q24/MYC, gains of 12q, and losses/CNN-LOH of 16p/CREBBP but with different frequencies. The number and the type of CNAs observed, as well as the mutational landscape in t(14;18)− FL in this study differs clearly from both NMZL and PTFL.
In summary, t(14;18)− FL is genetically a heterogeneous disorder with genetic features that differ from cFL, PTFL, and NMZL. The most distinct genetic alteration is the presence of STAT6 mutations, which usually co-occurred with TNFRSF14 and/or CREBBP alterations, revealing alternative oncogenic pathways. The genetic alterations seem to influence the morphology of t(14;18)− FL (follicular vs diffuse). Moreover, CD23 expression in t(14;18)− FL seems to be secondary to activating STAT6 mutations. Nevertheless, there is a group of t(14;18)− FL with low genomic complexity that warrants further study.
All copy number array files have been deposited in the Gene Expression Omnibus database (accession number GSE80103). The sequencing data are deposited in the European Nucleotide Archive (accession number PRJEB38853).
Acknowledgements
The authors thank Sema Colak, Esther Kohler, Claudia Hermann, and Noelia Garcia for excellent technical assistance and the IDIBAPS Genomics Core Facility.
This work was supported, in part, by grants from the Deutsche Forschungsgemeinschaft (DFG; QU144/1-1 [L.Q.-M.], FE597/4-1 [F.F.], and QU144/1-1 [I.M.]), Fondo de Investigaciones Sanitarias Instituto de Salud Carlos III (Miguel Servet Program CP13/00159 and PI15/00580, [I.S.]). J.E.R.-Z. was supported by a fellowship from the Generalitat de Catalunya Agencia de Gestión de la Universidad y la Investigación (AGAUR) FI-DGR 2017 (2017 FI_B01004). E.C. is an Academia Researcher of the “Institució Catalana de Recerca I Estudis Avançats” (ICREA) of the Generalitat de Catalunya. This work was partially developed at the Centro Esther Koplowitz, Barcelona, Spain. I.S. is supported by Acció Instrumental d’Incorporació de Científics i Tecnòlegs Plan de Investigación e Innovación en Salud (PERIS) 2016 (SLT002/16/00336) from the Generalitat de Catalunya.
Authorship
Contribution: I.S. and L.Q.-M. conceived and designed the study, supervised the experimental work, analyzed the data, and wrote the manuscript; D.N. and J.E.R.-Z. performed genetic analysis, interpreted the data, and helped write the manuscript; J.E.R.-Z. and G.C. performed bioinformatics analysis; I.M., J.S., F.O., S.M., and J.S.-V. performed genetic analyses and helped interpret the data; I.B. supervised the genetic analyses and helped interpret the data; B.M. performed and interpreted the FISH analysis; D.C. performed and interpreted some clonality analyses; C.E., B.G.-F., V.S., C.L.-M., L.L., S.D., A.C., C.C.-B., F.F., E.C., E.S.J., and L.Q.-M. contributed patients, reviewed their cases, and provided clinical information; E.C., B.G.-F., O.B., E.S.J., and L.Q.-M. performed the pathological review; and E.C., E.S.J., and F.F. analyzed the data and assisted in writing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Leticia Quintanilla-Martinez, Institute of Pathology, University Hospital Tübingen, Liebermeisterstr 8, 72076 Tübingen, Germany; e-mail: leticia.quintanilla-fend@med.uni-tuebingen.de; and Itziar Salaverria, Institut d´Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Rosselló 153, 08036 Barcelona, Spain; e-mail: isalaver@clinic.cat.

![Histologic features of t(14;18)−FL. (A-F) Case FL7, group B2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E]; original magnification, ×50). (B) The follicles are BCL2− with the 100/D5 clone (immunoperoxidase; original magnification, ×50). (C) The follicles are CD20+. (D) The tumor cells are CD23+. (E) MIB1 stain demonstrates low proliferation within the follicles without polarization. (F) The tumor cells are also negative with the alternative BCL2 antibody clone E17 (C-F; immunoperoxidase; original magnification, ×100). (G-M) Case FL11, group A1. (G) Inguinal LN with atypical lymphoid proliferation with follicular (right side) and diffuse (left side) growth pattern (H&E; original magnification, ×25). (H-I) The cells are negative with the BCL2 100/D5 clone and the E17 clone. (J-K) The tumor cells are CD10+ in the follicular and diffuse areas. (L-M) CD23 is strongly expressed in the follicular and diffuse areas (H-M; immunoperoxidase; original magnification, ×100).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/22/10.1182_bloodadvances.2020002944/2/m_advancesadv2020002944f1.png?Expires=1770622547&Signature=Z4ZEmNRRUP-5iQI7tCqYyzW2TgXO5RpZD14xzQWWUOKLD76IYz1kSPuADxNRYlSNiwyCDV62BoZ8WpCtp0CI1ucvb4hiStl~RgihyZzr5hzXxs50nE5S4-i~5~opoAj0ci1uPzQ4acXLKLCXtqw0bAtgAaXgW0xH7HHOn4w1~naWgRs5jEtQQUxjIvWRMRL5lwIQoW7o9SOKVqs9JjMYLhyegUFBhhvnaioXr2lS9nsA7~aRdDdSut1BpoNIdovEMTeJhH6ekSogx2C5G50WmKc2SCCFhIMJbDGaRItSSnWBXh~axoVjPP~kHfxsJEnVN3LBcxr1SOYk7ggxHw536w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Morphological features and CN comparison of t(14;18)−FL BCL6-break positive vs BCL6-break negative cases. (A-H) Case FL5, group A2. (A) Axillary LN with effaced architecture by an atypical lymphoid infiltration with follicular growth pattern. The follicles are back to back without well-defined mantle zones (hematoxylin and eosin [H&E] stain; original magnification, ×50). (B) Higher magnification demonstrates that the infiltrate is mainly composed of centrocytes with a few scattered centroblasts (H&E stain; original magnification, ×400). (C) The tumor cells are CD20+ (immunoperoxidase, original magnification, ×50). (D) BCL2 is negative in the tumor cells but positive in the residual mantle zones and reactive T cells. (E) The tumor cells are BCL6+. FISH analysis using a BCL6 break-apart assay (inset) shows a signal constellation of 1 colocalized signal (yellow arrow) and 1 split signal (red and green arrows) consistent with gene rearrangement. Tumor cells are CD10+ (F), whereas the residual follicular dendritic cells are CD23+ (G). Note that the tumor cells are CD23−. (H) MIB1 shows a low proliferation rate without polarization in the germinal centers (D-H; immunoperoxidase; original magnification, ×100). (I) Comparative plot of CN and CNN-LOH between 9 BCL6 break positive cases and 35 BCL6 break negative cases. The only significantly different region is indicated in the plot, and the color denotes the enriched group (Fisher’s exact test; adjusted P < .05; minimum 3 cases).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/22/10.1182_bloodadvances.2020002944/2/m_advancesadv2020002944f6.png?Expires=1770622547&Signature=jJNzhSydBBP33Tlo-B12k799ZsfXqgUeaBt0VME2OOSlk-iFwhsJjEkce~KjDCVqLwHtyh8QBkWxaZMBrnPgLhZhtHUsXfQv9G9a33HM3udl54dfvTK9sNIGntS5R3Zgj1AVXLBCN8x1v1xsq17LwJ-2O-4c4okDMj58EAU8674Sm6OgGlsbV3QF6Er8SWrGr2aRW4mALdxtxzdQWEXsZ9tlo~bzfCcm2jnWWsrFyCGqm7r4EZv5vmB9ExtIgZkmxf~QzAce-SDTH82XJCBZXLufejxS-FGD6fEAjD2H~w1X-OQ-OuCFZ-WS~eRlehTO39pDwHG7fMpUOirppJ7IWw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)

