Key Points
Distinctive circRNA expression profiles are associated with recurrent mutations and clinical features of CN-AML patients.
Individual circRNAs are associated with outcome and are functionally relevant in CN-AML.

Abstract
Circular RNAs (circRNAs) are noncoding RNA molecules that display a perturbed arrangement of exons, called backsplicing. To examine the prognostic and biologic significance of circRNA expression in cytogenetically normal acute myeloid leukemia (CN-AML), we conducted whole-transcriptome profiling in 365 younger adults (age 18-60 years) with CN-AML. We applied a novel pipeline, called Massive Scan for circRNA, to identify and quantify circRNA expression. We validated the high sensitivity and specificity of our pipeline by performing RNase R treatment and RNA sequencing in samples of AML patients and cell lines. Unsupervised clustering analyses identified 3 distinct circRNA expression–based clusters with different frequencies of clinical and molecular features. After dividing our cohort into training and validation data sets, we identified 4 circRNAs (circCFLAR, circKLHL8, circSMC1A, and circFCHO2) that were prognostic in both data sets; high expression of each prognostic circRNA was associated with longer disease-free, overall, and event-free survival. In multivariable analyses, high circKLHL8 and high circFCHO2 expression were independently associated with better clinical outcome of CN-AML patients, after adjusting for other covariates. To examine the biologic relevance of circRNA expression, we performed knockdown screening experiments in a subset of prognostic and gene mutation–related candidate circRNAs. We identified circFBXW7, but not its linear messenger RNA, as a regulator of the proliferative capacity of AML blasts. In summary, our findings underscore the molecular associations, prognostic significance, and functional relevance of circRNA expression in CN-AML.
Introduction
Aberrations in the noncoding transcriptome are emerging as important molecular mechanisms that contribute to cancer pathogenesis.1-3 The advent of next-generation sequencing has enabled the global, in-depth study of noncoding RNA species and has intensified the interest in their role in health and disease. Circular RNAs (circRNAs) are a class of noncoding, single-stranded RNA molecules that form covalently closed circles and are characterized by a perturbed arrangement of exons caused by aberrant splicing (referred to as backsplicing).4 CircRNAs were first identified almost 5 decades ago in plants, protozoans, and viruses5-7 and subsequently in mammals.8-10 Additional studies revealed a number of features that distinguish circRNAs from linear protein-coding RNA transcripts, such as the lack of 5′ or 3′ ends, the lack of polyadenylated tails, and increased stability and extended half-life.11 Although not yet fully elucidated, the biogenesis of circRNAs has been shown to depend on the spliceosome machinery,12-14 the presence of repeat sequences in the genome,9,10 and RNA binding proteins.15-17 The application of total RNA sequencing (RNA-seq) and novel bioinformatic approaches, designed to detect backspliced junctions, has uncovered the expression of a high number of circRNAs in humans, mice, metazoans, and plants.18 Recent reports have indicated that circRNAs are produced from ∼10% of all expressed genes, that a subset of them is evolutionarily conserved, and that circRNAs are often expressed in a tissue-specific manner.4,19,20 Although initially considered to represent inherent errors of the splicing process,2,8 recent studies have unraveled the biologic relevance of circRNAs.21-25
With regard to the functional significance of circRNAs, recent studies have demonstrated that circRNAs can sequester microRNAs (miRs) and inhibit their function. Two examples of such circRNAs are CDR1as/ciRS-7, which contains repetitive binding sites that capture miR-7,11,21 and circular SRY, which captures miR-138.11 Although other circRNAs have been proposed to function in the same manner,25-28 bioinformatic studies have indicated that only a small fraction of circular RNAs contain target sequences for miRs and can function as miR regulators.29,30 CircRNAs have also been shown to interact with RNA binding proteins or RNA polymerases and regulate gene transcription.31 In addition, recent studies have suggested that individual circRNAs can interact with ribosomes and be translated into peptides.32-36 However, the functional relevance and mechanism of action of the majority of the identified circRNAs remain unknown.
In cancer, expression levels of individual circRNAs have been identified as candidate biomarkers of disease progression.37-39 With regard to acute myeloid leukemia (AML), it was recently reported that chromosomal translocations, such as t(15;17)/PML-RARA or t(9;11)/MLLT3-KMT2A, give rise to fusion circRNAs that contribute to cellular transformation and survival of the leukemic blasts.40 However, the prognostic and functional significance of circRNA expression in AML has not been extensively studied in large patient cohorts. Herein, we performed total RNA-seq in younger adults (age <60 years) with de novo cytogenetically normal AML (CN-AML) and applied a novel bioinformatic algorithm to identify and quantify circRNAs. Our goals were to determine whether circRNA expression is associated with clinical and molecular features of CN-AML patients and whether circRNAs have functional relevance in this disease.
Patients, materials, and methods
Patients
Pretreatment bone marrow (BM) or blood samples were obtained from 365 younger adult patients (age 17-59 years) with de novo CN-AML. All analyzed patients were treated with cytarabine/anthracycline-based frontline chemotherapy on Cancer and Leukemia Group B (CALGB)/Alliance trials and were alive 30 days after initiation of treatment. No patient received allogeneic stem cell transplantation in first complete remission. Details regarding treatment protocols are provided in the supplemental Data. All patients were also enrolled onto companion protocols CALGB 8461 (cytogenetic studies), CALGB 9665 (Leukemia Tissue Bank) and CALGB 20202 (molecular analyses) (registered at www.clinicaltrials.gov as #NCT00048958 [CALGB 8461], #NCT00899223 [CALGB 9665] and #NCT00900224 [CALGB 20202]). All patients provided written informed consent, and all study protocols were in compliance with the Declaration of Helsinki and approved by institutional review boards.
Cytogenetic and gene mutation analyses
Cytogenetic analyses were performed in CALGB/Alliance-approved institutional laboratories and results confirmed by central karyotype review.41 The diagnosis of normal karyotype was based on the absence of clonal chromosome abnormalities in ≥20 metaphases obtained from BM samples subjected to 24- to 48-hour unstimulated cultures.41
Targeted amplicon sequencing using the Miseq platform (Illumina) was used to analyze DNA samples for the presence of gene mutations that are established prognosticators in CN-AML (ie, mutations in the ASXL1,42 DNMT3A [R882 and non-R882],43 IDH1, IDH2 [R140 and R172],44 NPM1,45 RUNX1,46 TET2,47 and WT1 genes48 and FLT3–tyrosine kinase domain [FLT3-TKD] mutations49 ) or that occur recurrently in CN-AML (ie, SF1, SF3A1, SF3B1, SRSF2, U2AF1, U2AF2, and ZRSR2), as described previously.50,51 A variant allele frequency of ≥10% was used as the cutoff to distinguish between mutated vs wild-type alleles. The presence of biallelic mutations in the CEBPA gene and FLT3–internal tandem duplications (FLT3-ITDs) were evaluated using Sanger sequencing and fragment analysis, respectively, as previously described.52,53
Transcriptome analyses
RNA samples of all patients (N = 365) were analyzed with RNA-seq (after depletion of ribosomal and mitochondrial RNA) using the Illumina HiSeq 2500 platform. Details regarding library generation protocols and sequencing are provided in the supplemental Data. To detect and quantify circRNA expression, we used a novel bioinformatic pipeline, called Massive Scan for circRNAs (MScircRNA), the features of which are presented in the Results section and in supplemental Data.
To determine the status of patients (ie, high vs low expressers) with regard to prognostic expression markers (ie, expression of the BAALC,54 MN1,55 and ERG56 genes and expression of miR-181a57 and miR-15558 ), the median values of normalized RNA-seq reads were used as the cutoff. With regard to miR-3151,59 patients expressing this miR were compared with those who did not. Data from small RNAs (miR-181a, miR-155, and miR-3151) were obtained using small RNA-seq.
Statistical analyses
Clinical end point definitions are given in the supplemental Data. Baseline demographic, clinical, and molecular features were compared between the training and validation sets, circRNA-expression based clusters, and low and high circRNA expressers using the Kruskal-Wallis and Fisher’s exact tests for continuous and categorical variables, respectively. The estimated probabilities of disease-free (DFS) and overall survival (OS) were calculated using the Kaplan-Meier method, and the log-rank test evaluated differences between survival distributions. Cox proportional hazards models were used to calculate hazard ratios (HRs) for DFS, OS, and event-free survival (EFS). Multivariable proportional hazards models were constructed for DFS, OS, and EFS using a backward selection procedure.60 Variables significant at α = 0.20 from the univariable analyses were considered for multivariable analyses (supplemental Data provides variables considered in model inclusion). For the time-to-event end points, the proportional hazards assumption was checked for each variable individually. All statistical analyses were performed by the Alliance Statistics and Data Center using SAS 9.4 and TIBCO Spotfire S+ 8.2 software. For laboratory in vitro experiments, 2-tailed Student t tests were performed. P < .05 was considered significant.
Results
MScircRNA: a novel pipeline for the detection and quantification of circular RNAs
To detect and quantify bona fide circRNAs, we developed a novel computational method called MScircRNA. As a starting point, we assembled a list with all backsplicing events detectable in our RNA-seq data set. The list was further filtered for backsplicing events compatible with perturbed arrangement of exons of the same gene. We compiled a FASTA file containing the sequences of the putative backsplicing junctions, as well as those annotated in the circBase database61 (supplemental Table 1). To quantify the expression levels of the detected circRNAs, we removed all linear reads and aligned the remaining reads to the curated list of candidate circRNA sequences. Fragments per kilobase pair of transcript per million mapped reads (FPKMs) were obtained by normalizing the fragment counts against the sequencing space across the backsplicing junction (supplemental Figure 1). Additional details regarding MScircRNA are provided in the supplemental Data.
Biochemical validation of the MScircRNA pipeline
To experimentally test our novel pipeline, we performed a sequencing experiment using samples from 7 CN-AML patients and 3 AML cell lines (Eol-1, OCI-AML3, and K-562) that were either mocktreated or treated with the RNase R exonuclease. RNase R preferentially degrades linear transcripts but has minimal effect on circularized transcripts and can therefore be used to differentiate between true circular RNAs and sequencing artifacts.62,63
We generated circRNA profiles of the mock and RNase R–treated samples using MScircRNA and identified 3031 circRNAs that were robustly expressed (FPKMs ≥1) in the 10 samples (supplemental Table 2). We found that RNase R treatment led to a significant FPKM increase for 2380 (78.5%) of the 3031 predicted circRNAs with respect to the untreated samples. Only 1 candidate circRNA (SNORD10) had significantly lower FPKMs in RNase R–treated AML samples (<0.01%). Regarding the remaining 650 circRNAs in which difference in expression between the 2 groups did not reach statistical significance, the trend was still that of enrichment because of resistance to RNase R for all except 10 circRNAs (10 [1.5%] of 650). Finally, in the case of 2323 circRNAs for which a related linear transcript could be detected and quantified, we calculated the ratio of the circular transcript FPKMs to the total FPKMs for the gene (circular FPKMs + linear FPKMs; ie, the circular fraction [CF]). It was recently proposed that the CF can be used as a parameter to distinguish circRNAs with functional significance from transcriptional noise.30 We found that for 2321 of these circRNAs, RNase R treatment increased the calculated CF (supplemental Table 3; supplemental Figure 2A-B). These results indicate that a vast majority of the circRNAs identified by MScircRNA were true circularized RNA transcripts and not artifacts of sequencing.
Circular RNA expression in AML and association with host linear gene expression
To study the prognostic and biologic significance of circRNA expression in CN-AML, we conducted analyses in a cohort of 365 younger CN-AML patients using MScircRNA (Table 1 lists the patients’ clinical features; supplemental Table 4 lists the RNA-seq characteristics). First, we detected and measured a total of 34 269 candidate circRNAs. To focus on the circular transcripts that were robustly and differentially expressed among the CN-AML samples, we applied stringent criteria. We retained the circRNAs, which were expressed above a threshold level (FPKM ≥1) and displayed variance of at least a 1.5-fold change in either direction from their corresponding median expression value. Additionally, we selected the circRNAs that had a CF >0.1 (which is the CF ratio threshold proposed to detect bona fide true circRNAs).30 These combined filters yielded a curated list of 180 circRNAs that were used in additional analyses (supplemental Table 5).
Associations between circRNA expression and molecular features of CN-AML patients
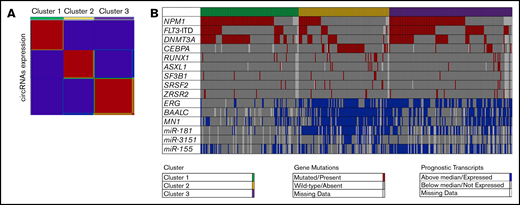
To examine whether distinctive patterns of circRNA expression were present in the data set of younger adults with CN-AML, we performed unsupervised clustering analysis. Specifically, we used a consensus nonnegative matrix factorization method to evaluate the segregation of the patients in circRNA-based clusters, according to circular expression. Cophenetic correlation coefficients indicated that the separation of patients into 3 distinct clusters generated the most robust consensus clustering (Figure 1A). Cluster 1 consisted of 115 patients, cluster 2 of 106, and cluster 3 of 144 (Figure 1B).
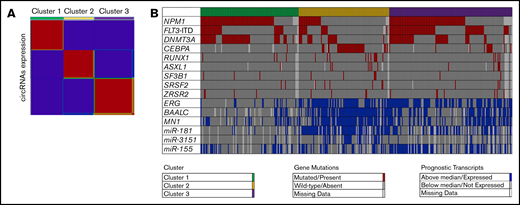
Unsupervised clustering of CN-AML patients based on expression of circRNAs. (A) Heatmap of circRNA expression in the 3 distinct circRNA expression–based clusters. Red indicates high and blue low expression. (B) Distribution of prognostic molecular features (ie, gene mutations, gene expression, and miR expression) across the 3 distinct circRNA expression–based clusters of younger adult patients with CN-AML.
Unsupervised clustering of CN-AML patients based on expression of circRNAs. (A) Heatmap of circRNA expression in the 3 distinct circRNA expression–based clusters. Red indicates high and blue low expression. (B) Distribution of prognostic molecular features (ie, gene mutations, gene expression, and miR expression) across the 3 distinct circRNA expression–based clusters of younger adult patients with CN-AML.
Overall, there were significant differences with regard to the distribution of clinical and molecular features among patients in the 3 circRNA-based clusters. Patients in cluster 1 were older than patients in clusters 2 and 3 (P = .02). Furthermore, cluster 1 patients had higher white blood cell (P < .001) and platelet counts (P < .001) but lower percentages of blasts in blood (P < .001) than patients in the other 2 clusters. Patients in cluster 1 were also more likely to present with extramedullary manifestations of their disease (P = .005). With regard to recurrent prognostic gene mutations, cluster 3 was enriched for the presence of FLT3-ITD. Concerning composite genotypes, cluster 3 patients were more likely to harbor the FLT3-ITD/NPM1–mutated genotype (Figure 1B). Patients in cluster 1 showed enrichment for NPM1 and DNMT3A mutations. The FLT3 wild-type/NPM1-mutated genotype was more frequent in patients in cluster 1. Cluster 2 showed a strong enrichment for the presence of mutations in transcription factors, such as biallelic CEBPA and RUNX1 mutations. NPM1 mutations, DNMT3A mutations, and FLT3-ITD were markedly underrepresented in cluster 2 (Table 2; Figure 1B).
The patients in the circRNA clusters also showed notable differences in expression levels of messenger RNAs (mRNAs) and miRs prognostic in CN-AML. Patients in cluster 3 were more frequently high expressers of ERG and miR-155, and those in cluster 2 of ERG BAALC, MN1, miR-181a, and miR-3151. In contrast, patients with low expression of prognostic mRNAs and miRNAs were significantly overrepresented in cluster 1 (Table 2; Figure 1B).
Prognostic significance of circRNA expression in younger adults with CN-AML
To evaluate the prognostic significance of circRNA expression in CN-AML, we randomly divided our cohort of younger CN-AML patients into a training set, used for exploratory analysis (n = 254), and a validation set (n = 111). There were no significant differences in clinical and molecular features between the 2 groups, except for percentages of blasts in blood (higher in training set patients; P = .03), frequency of FLT3-TKD mutations (more frequent in the training set; P = .02), and ERG (P = .01) and BAALC (P = .002) expression levels (for both genes, training set patients were more frequently high expressers; supplemental Table 6).
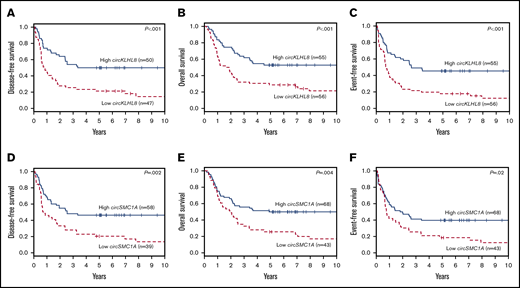
Next, we evaluated the association of circRNA expression with EFS in the training set using univariate Cox regression. EFS was chosen because it is a composite outcome end point that comprehensively evaluates response to chemotherapy, probability of relapse, and probability of survival. Twelve circRNAs were found to be significantly associated with EFS in the training set (threshold P < .05; supplemental Table 7). Four of these 12 circRNAs were also significantly associated with clinical outcome in the validation data set (circCFLAR, circKLHL8, circSMC1A, and circFCHO2). circKLHL8 and circSMC1A showed the strongest association with clinical outcome; patients with high circKLHL8 expression had longer DFS, OS, and EFS than patients with low circKLHL8 expression (P < .001 for all 3 end points; Figure 2A-C). Patients with high circSMC1A had longer DFS (P = .002), OS (P = .004), and EFS (P = .02) than patients with low circSMC1A expression (Figure 2D-F). Similarly, patients with either high circFCHO2 expression or high circCFLAR expression had better outcomes than those of patients with low circFCHO2 or circCFLAR expression, respectively (supplemental Figure 3A-F). We did not detect an association between circRNA expression and complete remission rates in either of the data sets (data not shown).
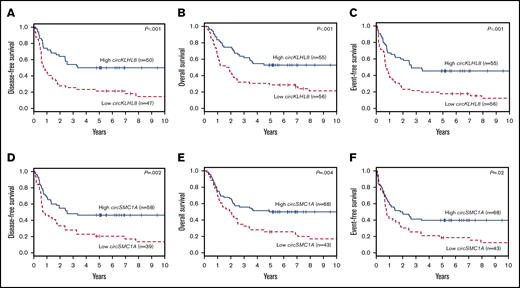
Clinical outcome of 111 younger adult patients with CN-AML in the validation set with either high or low expression of individual circRNAs. DFS (A), OS (B), and EFS (C) of high vs low expressers of circKLHL8. DFS (D), OS (E), and EFS (F) of high vs low expressers of circSMC1A.
Clinical outcome of 111 younger adult patients with CN-AML in the validation set with either high or low expression of individual circRNAs. DFS (A), OS (B), and EFS (C) of high vs low expressers of circKLHL8. DFS (D), OS (E), and EFS (F) of high vs low expressers of circSMC1A.
Validation of the prognostic circRNA expression status of CN-AML
Before further studying the circRNAs, which were found to be associated with clinical outcome of younger adult CN-AML patients, we sought to validate our circRNA expression measurements and the distinction of patients in high vs low expressers with additional transcriptome profiling methods. To this end, we designed real-time quantitative polymerase chain reaction (RT-qPCR) assays that target the backsplicing regions of circRNA transcripts and can quantify their expression levels. We then measured circRNA expression in subsets of patients who had been classified as high or low expressers of the respective prognostic circRNAs based on MScircRNA analysis. There was overall high concordance between the MScircRNA and RT-PCR measurements in these patients, as indicated by the Pearson’s correlation coefficient values (Pearson’s r, 0.94-0.99; supplemental Figure 4A-D).
Associations of prognostic circRNAs and pretreatment clinical and molecular features of CN-AML patients
Next, we investigated whether the expression of the prognostic circRNAs in CN-AML patients were associated with distinctive clinical or molecular features. Patients with high circKLHL8 expression had higher platelet counts (P = .05) and lower percentages of blasts in blood (P = .002) and BM (P = .05) than patients with low circKLHL8 expression (supplemental Table 8). High circKLHL8 expressers were also more likely to have FLT3-TKD (P = .05) and less likely to have FLT3-ITD (P < .001) or high expression of miR-155 (P < .001) or ERG (P < .001; supplemental Table 8). Patients with high circSMC1A were more likely to be female (P = .002) and less likely to have FLT3-ITD (P = .04) than patients with low circSMC1A expression (supplemental Table 9). The clinical and molecular features associated with high expression of circCFLAR and circFCHO2 are provided in supplemental Tables 10 and 11, respectively.
Association of prognostic circRNA expression with corresponding linear transcripts
To ensure that our observations on prognostic circRNAs were not merely reflecting the prognostic significance of the corresponding linear transcripts, we examined the correlation of circular and linear RNA expression for the 4 circRNAs with validated prognostic significance. With the exception of circFCHO2, we found the correlation between circular and linear transcripts to be weak (range of Pearson’s r, −0.05 to 0.29). In keeping with these findings, linear CFLAR, KLHL8, and SMC1A mRNAs were not prognostic in the validation set of younger adults with CN-AML. In contrast, circFCHO2 and linear FCHO2 were moderately correlated in the validation set (Pearson’s r, 0.69). High linear FCHO2 expression also was associated with better clinical outcome and longer DFS, although not as strongly as circFCHO2 expression (supplemental Figure 5A-L).
Multivariable analyses
To evaluate the prognostic significance of circRNAs in the context of established prognostic markers, we constructed multivariable models, concomitantly analyzing expression of the 4 prognostic circRNAs. With regard to DFS, high circKLHL8 expression remained significantly associated with longer DFS (HR, 0.53; P = .02) after adjusting for presence of FLT3-ITD and MN1 expression status. High circKLHL8 (HR, 0.53; P = .01) and high circFCHO2 (HR, 0.45; P = .002) were significantly associated with longer OS after adjusting for MN1 expression status. Finally, high circKLHL8 and high circFCHO2 expression were significantly associated with longer EFS (HR, 0.46; P = .002 and HR, 0.56; P = .02, respectively) after adjusting for the presence of DNMT3A mutations and MN1 expression status (Table 3).
Molecular pathways associated with expression of prognostic circRNAs
To gain mechanistic insights and detect the molecular pathways associated with the expression levels of the prognostic circRNAs, we performed transcriptome and molecular pathway analyses and compared patients with high with those with low expression of the prognostic circRNAs.
We found circCFLAR and circKLHL8 expression status to be associated with a high number of differentially expressed mRNAs (supplemental Tables 12 and 13, respectively). High circCFLAR expression was associated with increased abundance of transcripts involved in intracellular signaling (SGK1 and RGS2) and regulation of gene expression (SMAD7, KMD7A, and ID2). In pathway analysis, high circCFLAR expression was found to be associated with immune system activation, leukocyte chemotaxis, and cytokine excretion (supplemental Table 14). High circKLHL8 was associated with increased expression of cell cycle, apoptosis, or immune response regulators (CDKN1, CDKN2, BCL6, and TLR4) as well as transcription factors involved in macrophage differentiation (CEBPD and CEBPB). In pathway analysis, high circKLHL8 expression was associated with activation of the differentiation and apoptosis pathways, as well as with cytokine production and secretion (supplemental Table 15). CircFCHO2 and circSMC1A expression were not associated with distinct gene expression patterns.
Because circRNAs have been shown to function as decoys that sequester and inhibit the function of miRs, we used an in silico approach and publicly available databases (miRBase64 and TargetScan65 ) to examine whether the prognostic circRNAs contained miR-binding regions. We did not identify repeat regions that could function as miR-sequestering sites in the nucleotide sequence of any of the prognostic circRNAs.
Biologic significance of circRNA expression in AML
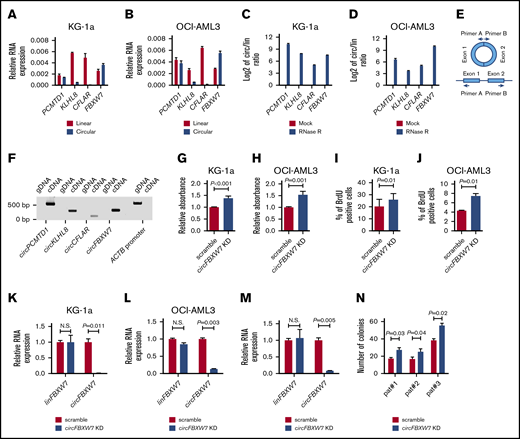
To evaluate the biologic significance of circRNA expression in AML, we performed loss-of-function in vitro experiments focusing on 4 circRNAs (circPCMTD1, circKLHL8, circCFLAR, and circFBXW7) that were either prognostic or associated with established, recurrent gene mutations in this disease. Selection of candidate circRNAs was also based on our predicted capacity to generate oligonucleotides that would target the circRNAs without perturbing the expression of the corresponding linear transcripts. We first profiled the expression of these 4 circRNAs and the corresponding linear RNA transcripts with RT-qPCR assays in 2 AML cell lines (KG-1a and OCI-AML3). In both cell lines, we found that circKLHL8 and circCFLAR represented small fractions of the total amount of KLHL8 and CFLAR transcripts, whereas circFBXW7 and circPCMTD1 were expressed at similar or higher levels than linear PCMTD1 and linear FBXW7, respectively (Figure 3A-B). We confirmed the circular nature of the transcripts we studied by demonstrating their resistance to RNase R and the subsequent increase of the circular/linear transcript ratio upon RNase R treatment (Figure 3C-D). In addition, we performed PCR experiments using divergent primers that would bind to DNA regions adjacent to the backsplicing sites of the circRNAs (Figure 3E). These assays generated amplicons on reverse transcribed RNA (ie, complementary DNA) but not genomic DNA of KG-1a cells (Figure 3F; supplemental Table 16). Cloning of the amplicons into bacterial vectors and Sanger sequencing confirmed the RNAseq-predicted sequences of the circular transcripts and showed that they consisted of fused exons.
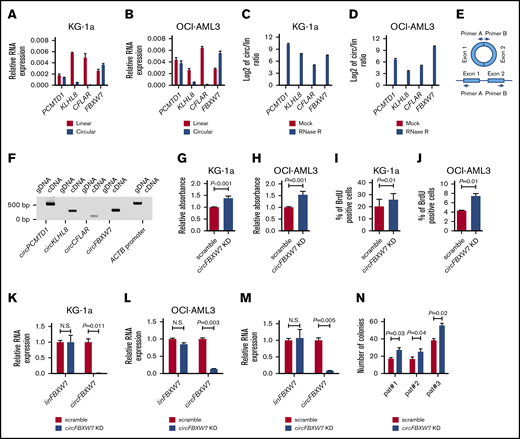
Functional significance of circRNA expression in AML. (A-B) Relative expression of linPCMTD1, circPCMTD1, linKLHL8, circKLHL8, linCFLAR, circCFLAR, linFBXW7, and circFBXW7 in KG-1a (A) and OCI-AML3 (B) cells. (C-D) Circular-to-linear (circ/lin) ratio of the PCMTD1, KLHL8, CFLAR, and FBXW7 transcripts in mock vs RNase R–treated samples of KG1a (C) and OCI-AML3 (D) cells. The log2 value of the circ/lin ratio is depicted in the figures. (E) Schematic description of the PCR experiments with divergent primers. The reverse primer (primer A) binds to the 5′ end of exon 1 and the forward primer (primer B) binds to the 3′ end of exon 2. With this design, amplicons can be generated after hybridization of the primers on circular RNA transcripts but not on genomic DNA. (F) circPCMTD1, circKLHL8, circCFLAR, and circFBXW7 PCR results using complementary DNA and genomic DNA of KG-1a cells. The integrity of the DNA was tested with primers specific for the ACTB DNA promoter region. (G-H) Proliferation analyses of scramble vs circFBXW7 KD-treated KG-1a (G) and OCI-AML3 (H) cells, as measured by a colorimetric assay, based on degradation of the WST1 reagent. (I-J) Proliferation analyses of scramble vs circFBXW7 KD-treated KG-1a (I) and OCI-AML3 (J) cells, based on BrdU incorporation. (K-M) Relative RNA expression of linFBXW7 and circFBXW7 in scramble vs circFBXW7 KD-treated KG-1a cells (K), OCI-AML3 cells (L), and AML patient blasts (M). (M) Samples of 1 patient are depicted as an example. (N) Colony-forming unit assays in methylcellulose-based media with scramble vs circFBXW7 KD-treated AML patient blasts of 3 different patients. N.S., not significant.
Functional significance of circRNA expression in AML. (A-B) Relative expression of linPCMTD1, circPCMTD1, linKLHL8, circKLHL8, linCFLAR, circCFLAR, linFBXW7, and circFBXW7 in KG-1a (A) and OCI-AML3 (B) cells. (C-D) Circular-to-linear (circ/lin) ratio of the PCMTD1, KLHL8, CFLAR, and FBXW7 transcripts in mock vs RNase R–treated samples of KG1a (C) and OCI-AML3 (D) cells. The log2 value of the circ/lin ratio is depicted in the figures. (E) Schematic description of the PCR experiments with divergent primers. The reverse primer (primer A) binds to the 5′ end of exon 1 and the forward primer (primer B) binds to the 3′ end of exon 2. With this design, amplicons can be generated after hybridization of the primers on circular RNA transcripts but not on genomic DNA. (F) circPCMTD1, circKLHL8, circCFLAR, and circFBXW7 PCR results using complementary DNA and genomic DNA of KG-1a cells. The integrity of the DNA was tested with primers specific for the ACTB DNA promoter region. (G-H) Proliferation analyses of scramble vs circFBXW7 KD-treated KG-1a (G) and OCI-AML3 (H) cells, as measured by a colorimetric assay, based on degradation of the WST1 reagent. (I-J) Proliferation analyses of scramble vs circFBXW7 KD-treated KG-1a (I) and OCI-AML3 (J) cells, based on BrdU incorporation. (K-M) Relative RNA expression of linFBXW7 and circFBXW7 in scramble vs circFBXW7 KD-treated KG-1a cells (K), OCI-AML3 cells (L), and AML patient blasts (M). (M) Samples of 1 patient are depicted as an example. (N) Colony-forming unit assays in methylcellulose-based media with scramble vs circFBXW7 KD-treated AML patient blasts of 3 different patients. N.S., not significant.
For the knockdown (KD) experiments, we used RNase H-recruiting LNA-modified oligonucleotides (gapmers) that specifically targeted the backsplicing regions of the circRNAs (supplemental Table 17). Of the 4 tested circRNAs, depletion of circFBXW7 affected the phenotype of the leukemic cells. More specifically, circFBXW7 KD led to an increase in the proliferative capacity of KG-1a and OCI-AML3 cells as measured by WST1 degradation (P < .001 and P = .001, respectively; Figure 3G-H). In addition, cell proliferation analysis, using BrdU, confirmed an increase in the fraction of proliferating cells upon depletion of circFBXW7 in both KG-1a and OCI-AML3 cells (P = .01 in both cases; Figure 3I-J). RT-qPCR showed that the gapmers specifically targeted circFBXW7 without affecting the expression level of linear FBXW7 mRNA (Figure 3K-L).
To further study the functional significance of circFBXW7, we performed KD experiments in blasts of 3 AML patients. RT-qPCR confirmed that gapmers also induced specific depletion of circFBXW7 in the blasts, with no effect on the level of the linear transcript (Figure 3M). In all 3 patients, circFBXW7 KD generated a similar phenotype and led to an increase in the number of colonies formed by the blasts in methylcellulose-based assays (Figure 3N).
To evaluate whether miR sequestration could be the mechanistic basis of circFBXW7 function, we used a bioinformatics-based approach and searched the miRBase and TargetScan databases, as described in “Molecular pathways associated with expression of prognostic circRNAs.” There were no detectable miR-binding sites contained within the cirFBXW7 sequence, arguing against an miR-dependent mechanism of action for this circRNA.
To study the molecular pathways that are associated with the expression levels of circFBXW7, we performed transcriptome analysis in high vs low circFBXW7 expressers in our cohort of younger adults with CN-AML. We found that high expression of circFBXW7 was associated with a distinct gene expression signature and high expression of genes involved in the regulation of chromatin state and transcription (HIST1H4F, HIST1H2BG, HISTH2AE, and MLLT3) as well as key transcription factors and signal transduction mediators implicated in leukocyte activation and differentiation (IKZF2, AKT3, and RHOBT3; supplemental Table 18). Low expression of circFBXW7 was associated with increased expression of known regulators of stem cell properties, such as multiple Homeobox genes (HOXA2, HOXA7, HOXA9, HOXB3, and MEIS1) as well as genes involved in the WNT/β-catenin and NOTCH signaling pathways (PRICKLE1 and JAG1; supplemental Table 18; supplemental Figure 6). In pathway analysis, high expression of circFBXW7 was found to be associated with positive regulation of signal transduction and leukocyte differentiation (supplemental Table 19). In contrast, decreased expression of circFBXW7 was associated with the process of hemopoiesis and positive regulation of RNA biosynthesis (supplemental Table 20). Collectively, these data show that circRNA expression, in addition to its association with clinical outcome and prognostic mutations, also has biologic significance in CN-AML.
Discussion
CircRNAs are a novel class of noncoding RNA molecules that are gaining increasing recognition for their functional significance in health and disease.21-25 Although circRNAs have previously been reported to stem from chromosomal translocations that are recurrent in AML and to be involved in malignant transformation,40 their prognostic and biologic significance in AML has not been extensively studied.
In this work, we have profiled a large number of samples from CN-AML patients using ribosomal RNA-depleted RNA-seq protocols to capture RNA transcripts independent of polyadenylation status. Most of the available RNA-seq data sets from AML patients have been generated using polyadenosine tail-based selection, because this has been shown to be the most cost-effective approach for the study of protein-coding transcripts. In contrast to these data sets, our approach allows profiling of a wide variety of noncoding transcripts in addition to mRNAs. Thus, the availability of whole-transcriptome sequencing data of CN-AML patients with detailed clinical and molecular information is an important step toward understanding noncoding RNA biology and discovering novel biomarkers and therapeutic targets.
We studied the expression of circRNAs and their association with clinical features and outcome of patients with CN-AML and performed experiments to investigate whether circRNAs are functionally relevant in this disease. We developed a new algorithm (MScircRNA) to detect and quantify circRNA transcripts from RNA-seq. We further validated in vitro the performance of our pipeline by conducting analysis on samples treated with RNase R, the gold-standard method for identifying true circular transcripts. Altogether, our results confirmed that MScircRNA is a reliable and sensitive method for circRNA detection and quantification.
Global circRNA expression profiling classified CN-AML samples of our data set into 3 groups, each with distinctive enrichment of recurrent mutations and clinical features. By using a robust training/validation approach, we identified 4 circRNAs that were associated with outcome. In multivariable analyses, high expression of 2 of these circRNAs (circKLHL8 and circFCHO2) were independently associated with EFS, DFS, and OS after adjusting for other covariates.
Lastly, we conducted proof-of-principle experiments to evaluate the biologic role of circRNAs in AML. We performed a small-scale KD screening, focusing on 4 candidate circRNAs and using oligonucleotides, which targeted the circRNAs without perturbing the corresponding linear transcripts. Depletion of 1 of the candidate circRNAs, circFBXW7, affected the phenotype of the AML cell lines and primary AML samples that were tested and led to an increase in their proliferative capacity. On the basis of the lack of miR binding sites in circFBXW7, it is unlikely that these effects are mediated via sequestration of miRs. Although more experiments will be needed to understand how circFBXW7 regulates proliferation of the leukemic blasts, our current data lend support to the functional relevance of circRNA expression in AML.
In summary, we have performed the first comprehensive circRNA profiling of a large CN-AML patient cohort. We identified distinctive clusters of circRNA expression, which are associated with recurrent mutations and clinical features of CN-AML patients. In addition, we provide evidence that 1 of the analyzed circRNAs, circFBXW7, acts as a tumor suppressor in AML.
Presented in abstract form at the 23rd Annual Meeting of the European Hematology Association, Stockholm, Sweden, 14-18 June 2018.
Data sharing requests can be e-mailed to the corresponding author, Ramiro Garzon (ramiro.garzon@osumc.edu).
Acknowledgments
The authors thank the Alliance Hematology Malignancy Biorepository and the Alliance National Cancer Institute National Clinical Trials Network (NCTN) Biorepository and Biospecimen Resource for sample processing and storage services and Lisa J. Sterling and Christine Finks (The Ohio State University Comprehensive Cancer Center, Columbus, OH) for data management.
This work was supported by the National Cancer Institute (NCI), National Institutes of Health (NIH), under awards U10CA180821 and U10CA180882 (to the Alliance for Clinical Trials in Oncology), UG1CA233338, U10CA077658, U10CA180850, U10CA180861, CA140158, CA16058, and R35 CA197734. This work was also supported in part by the Leukemia Clinical Research Foundation, D. Warren Brown Foundation, and Pelotonia Fellowship Program. S.V. was supported by the Associazione Italiana Ricerca sul Cancro. R.G. was supported by a Scholar in Clinical Research award from the Leukemia & Lymphoma Society. The Alliance Hematology Malignancy Biorepository was supported by Washington University subcontract WU-15-398/WU-16-51, and the Alliance NCTN Biorepository and Biospecimen Resource was supported by NCI NIH award U24CA196171.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: D.P., S.V., M.Ś., C.D.B., and R.G. conceived and designed the study; D.P., S.V., D.N., K.M., C.D.B., and R.G. drafted the manuscript; D.P., S.V., D.N., M.Ś. and J.K. analyzed data; S.V. and M.Ś. contributed vital new analytical tools; A.P. and S.K. contributed vital new reagents; J.C.B., C.D.B., and R.G. obtained funding for this study; C.D.B. and R.G. supervised the study; and all authors participated in the acquisition, analysis, and interpretation of the data and in the critical revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ramiro Garzon, The Ohio State University Comprehensive Cancer Center, 460 West 12th Ave, Columbus, OH 43210; e-mail: ramiro.garzon@osumc.edu.