

Key Points
BCL-W is dispensable for the growth and survival of select BL- and DLBCL-derived cell lines.

Abstract
Dysregulated expression of BCL-2 family proteins allows cancer cells to escape apoptosis. To counter this, BH3-mimetic drugs that target and inhibit select BCL-2 prosurvival proteins to induce apoptosis have been developed for cancer therapy. Venetoclax, which targets BCL-2, has been effective as therapy for patients with chronic lymphocytic leukemia, and MCL-1–targeting BH3-mimetic drugs have been extensively evaluated in preclinical studies for a range of blood cancers. Recently, BCL-W, a relatively understudied prosurvival member of the BCL-2 protein family, has been reported to be abnormally upregulated in Burkitt lymphoma (BL), diffuse large B-cell lymphoma (DLBCL), and Hodgkin lymphoma patient samples. Therefore, to determine if BCL-W would be a promising therapeutic target for B-cell lymphomas, we have examined the role of BCL-W in the sustained growth of human BL- and DLBCL-derived cell lines. We found that CRISPR/CAS9-mediated loss or short hairpin RNA-mediated knockdown of BCL-W expression in selected BL and DLBCL cell lines did not lead to spontaneous apoptosis and had no effect on their sensitivity to a range of BH3-mimetic drugs targeting other BCL-2 prosurvival proteins. Our results suggest that BCL-W is not universally required for the sustained growth and survival of human BL and DLBCL cell lines. Thus, targeting BCL-W in this subset of B-cell lymphomas may not be of broad therapeutic benefit.
Introduction
BCL-W (BCL2L2) was first described as a BCL-2 family protein that, upon overexpression, could protect lymphoid and myeloid cells from apoptosis induced by cytokine deprivation and γ-irradiation.1 BCL-W–deficient mice were found to be phenotypically normal, with the exception of aberrant germ cell apoptosis in the adult testes, resulting in a lack of mature sperm.2,3 Because of the lack of overt developmental and health defects in BCL-W–deficient mice, BCL-W is one of the least studied members of the BCL-2 protein family. This family is characterized by the presence of 1 or more BCL-2 homology (BH) domains and includes both prosurvival and proapoptotic factors.4,5 The prosurvival proteins (BCL-W, BCL-2, MCL-1, BCL-XL, and BFL-1/A1) are responsible for binding and restraining the proapoptotic effectors (BAK and BAX). This prevents them from forming pores in the mitochondrial outer membrane and thus precludes initiation of the caspase cascade that causes demolition of cells undergoing apoptosis. The proapoptotic BH3-only proteins (BIM, BAD, BID, PUMA, BMF, BIK, HRK, and NOXA) can bind and sequester the prosurvival BCL-2 proteins, thereby unleashing BAK and BAX and allowing apoptosis to proceed. Additionally, certain BH3-only proteins (eg, BIM, PUMA, tBID) have been reported to directly activate BAX and BAK.6 The balance and interactions between prosurvival and proapoptotic BCL-2 family proteins within a cell determines its fate.
Defects in cell death regulation are tightly linked to tumor development and sustained survival of malignant cells; thus, evasion of apoptosis is a well-recognized hallmark of cancer.7 Upregulation of prosurvival BCL-2 family members, such as via somatically acquired amplifications of the genomic region containing the MCL-1 or BCL-X genes found in ∼10% to 15% or ∼5%, respectively, of diverse cancers,8 or loss of proapoptotic BH3-only proteins9,10 are commonly associated with malignant diseases. Genetic experiments revealed that cancer cells can display a dependence on 1 particular prosurvival BCL-2 protein for ongoing survival; multiple myeloma and Burkitt lymphoma (BL) cells are largely reliant on MCL-1,11,12 whereas chronic lymphocytic leukemia cells exhibit BCL-2 dependency.13 Accordingly, the development of BH3-mimetic drugs that can bind and inhibit specific prosurvival BCL-2 family proteins has been an intense area of study over the past decade,14,15 culminating in dozens of clinical trials and, ultimately, US Food and Drug Administration approval of the BCL-2 inhibitor venetoclax for the treatment of patients with chronic lymphocytic leukemia5,16,17 and acute myeloid leukemia.18,19 BH3-mimetic drugs targeting other prosurvival proteins are in various stages of development. Clinical trials commenced with MCL-1 inhibitors for certain B-cell malignancies, acute myeloid leukemia, and multiple myeloma.20 ABT-263/navitoclax, which targets BCL-2, BCL-XL, and BCL-W, as well as BCL-XL specific inhibitors, such as WEHI-539, were shown to potently kill diverse cancer-derived cell lines in culture and in vivo.21,22 However, on-target toxicity to platelets, which rely on BCL-XL for survival, has stalled the progression of these drugs in clinical trials.23,24
Recent reports have implicated a role for BCL-W in human cancers. It was shown that BCL-W is significantly overexpressed in a wide range of human B-cell lymphomas, including BL, diffuse large B-cell lymphoma (DLBCL), and Hodgkin lymphoma (HL) patient samples and cell lines.25-27 It has also been reported that loss of BCL-W delays lymphoma development in the Eμ-Myc transgenic mouse model of BL and, importantly, that BCL-W expression is essential for the sustained survival of MYC-driven human BL-derived cell lines.25 Finally, a CRISPR/CAS9 functional screen identified BCL-W as a factor rendering a range of human adenocarcinoma-derived cell lines resistant to BH3-mimetic drugs targeting BCL-2, MCL-1 or BCL-XL.28 Together, these studies indicate a previously underappreciated role for BCL-W in cancer and identify BCL-W as a potentially attractive anticancer drug target.
In light of these reports, we sought to independently validate a role for BCL-W in the survival of human BL and DLBCL cell lines. In contrast to a previous study,25 we found that BCL-W was not uniformly expressed at high levels across the BL and DLBCL cell lines examined. Notably, reduction of BCL-W expression using CRISPR/CAS9 gene editing or RNA interference in a panel of lymphoma cell lines did not sensitize them to apoptosis, even when these cells were treated with BH3-mimetic drugs targeting other prosurvival BCL-2 proteins.
Materials and methods
Cell culture
Ramos-BL and Raji-BL were kind gifts from Suzanne Cory, The Walter and Eliza Hall Institute of Medical Research (originally received from George Klein) and David Huang, The Walter and Eliza Hall Institute of Medical Research, respectively. The DLBCL cell lines U2932, HT, SUDHL-4, and SUDHL-5 were obtained from the Germany Collection of Microorganisms and Cell Cultures (DSMZ). All cell lines were authenticated by STR profiling at the Australian Genome Research Facility and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; MilliporeSigma), 100 U/mL penicillin, and 100 μg/mL streptomycin, and maintained at 5% CO2. HEK293T cells were cultured in Dulbecco’s modified Eagle medium (Thermo Fisher Scientific) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin, and maintained at 10% CO2. All cell lines were verified as mycoplasma free.
Western blotting
Total protein was extracted by lysis in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCL, 150 mM NaCl, 1% NP-40, 0.5% DOC, 0.1% sodium dodecyl sulfate) containing complete protease inhibitor cocktail (Roche) and quantified by Bradford assay (Bio-Rad). Between 10 μg to 25 μg of protein was run on 10% or 4% to 12% ν-polyacrylamide gel electrophoresis gels (Thermo Fisher Scientific) and then transferred onto a nitrocellulose membrane. Membranes were blocked in phosphate-buffered saline containing 0.1% Tween with 5% skim milk. Membranes were incubated in primary antibodies diluted in phosphate-buffered saline containing 0.1% Tween, 5% bovine serum albumin (MilliporeSigma) and 0.02% sodium azide. Antibodies are detailed in supplemental Table 1. Membranes were imaged by addition of Immobilon Forte Western HRP Substrate (Merck) on a ChemiDoc Imaging System (Bio-Rad). Quantification of western blots was performed using ImageJ (US National Institutes of Health). Data were normalized to the HSP70 loading control.
qRT-PCR
Total RNA was extracted using Trizol reagent (Thermo Fisher Scientific) and complementary DNA was synthesized using the SuperScript III First Strand Synthesis Kit (Thermo Fisher Scientific) with oligo(dT)20 primers, both methods according to the manufacturer’s instructions. Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis was performed in triplicate using TaqMan Gene Expression Assays (Applied Biosystems) on the ViiA7 Real-Time PCR System (Thermo Fisher Scientific), detailed in supplemental Table 2. Data were analyzed using QuantStudio software (Thermo Fisher Scientific). Messenger RNA (mRNA) levels were normalized to expression of the housekeeping gene HMBS.
CRISPR/CAS9 and shRNA constructs, virus production, and cell sorting
CRISPR/CAS9 gene editing was performed by serial infection using a lentiviral system composed of stably expressed CAS9, followed by doxycycline-inducible expression of single guide RNA (sgRNA), as described elsewhere.29 A nontargeting control sgRNA (sgNT) construct against mouse Bim exon 2 (5′-GACAATTGCAGCCTGCTGAG-3′)29 and sgRNA construct targeting human BCL-W exon 3 (sgBCL-W: 5′-GGAGTTCACAGCTCTATACG-3′)30 were used. For RNA interference, an inducible short hairpin RNA (shRNA) construct was designed by cloning of a BCL-W target sequence (shBCL-W: 5′-TGGCAGACTTTGTAGGTTATA-3′)31 into the lentiviral pF1HtUTG vector, as described.32 An shRNA construct targeting rat CD8 was used as a negative control.32 For both methods, lentivirus was produced by transient transfection into HEK293T cells using calcium phosphate precipitation according to published protocols.32 Infected cells were sorted for high expression of mCherry from the Cas9 expression vector, eGFP from the sgRNA vector, or eGFP from the shRNA vector on an Aria W flow cytometer (Becton Dickinson).
Induction of gene editing or RNA interference
Doxycycline hyclate (MilliporeSigma; #D9891) was added to tissue culture medium at a concentration of 1 μg/mL to induce sgRNA or shRNA expression. Doxycycline levels were topped up every 24 hours during sgRNA or shRNA induction. Cell viability was determined by staining with Annexin V-Alexa Fluor 647 and DAPI, and quantified by flow cytometry on an LSR IIW flow cytometer (Becton Dickinson). Live cells were deemed Annexin V and DAPI double-negative. For proliferation assays, cell number was determined using a Moxi Z cell counter (ORFLO).
Next-generation sequencing
DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s protocol and PCR amplified using sgRNA-specific primers and GoTaq Green Master Mix (Promega). PCR products were subjected to a second round of PCR using indexing primers, pooled, purified using Ampure XP beads (Beckman Coulter), and sequenced using the MiSeq platform (Illumina). Reads were mapped to the human BCL-W reference sequence using the Basic Local Alignment Search Tool (National Center for Biotechnology Information) and the proportion of mutations for each cell line were calculated in Microsoft Excel.
BCL-W target site primers were FWD 5′-GTGACCTATGAACTCAGGAGTCCGCAGTGGATGGAACTGGAA-3′ and REV 5′-CTGAGACTTGCACATCGCAGCGCCCTGGACTTTCACTTGCT-3′.
Drug treatment assays
Cells were plated in triplicate at 3 × 104 cells/well of flat-bottomed 96-well plates. ABT-199 (Active Biochem; #A-1231), ABT-737 (Active Biochem; #1002), S63845 (Active Biochem; #6044), or A-1331852 (AbbVie, provided by G. Lessene) in dimethyl sulfoxide (DMSO) were added to the specified concentration. Cell viability was determined at 24 hours, as previously described. Data were normalized to control cells treated with an equivalent amount of DMSO (vehicle). For assays in shRNA-induced cells, drug treatments were performed following 72 hours of shRNA induction in the presence of doxycycline.
Generation of single-cell gene-edited clones
Cells with high expression of Cas9 (mCherry) and BCL-W or NT sgRNA (eGFP) were sorted into round-bottomed 96-well plates at 1 cell per well on an Aria W flow cytometer. Twenty-four clones were expanded, sgRNA expression was induced with doxycycline, and cells were screened for disruption of BCL2L2 (encodes BCL-W) by western blotting and sequencing as described previously. Drug treatment assays were performed as previously. For low-serum experiments, clones were grown in RPMI-1640 medium supplemented with 1% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin for 72 hours before drug treatment.
Statistical analysis
GraphPad Prism software was used for statistical analyses. Two-tailed Student t tests were performed to compare between 2 data sets. One-way analysis of variance with Dunnett’s post hoc test was used to compare between more than 2 samples. Error bars represent the standard deviation (SD) of 3 independent experiments unless otherwise indicated.
Results
Characterization of human B-cell lymphoma lines
Six human B-cell lymphoma-derived cell lines were analyzed for expression of the prosurvival proteins BCL-W, BCL-2, BCL-XL, BFL-1, and MCL-1, along with proapoptotic BIM that is frequently epigenetically silenced in BLs,9 and the tumor suppressor TP53, which is mutated in ∼50% of human cancers.33 The selected cell lines included the BL lines Ramos-BL and Raji-BL, which were previously shown to be dependent on BCL-W for survival25 and the DLBCL lines U2932 (activated B-cell subtype), SUDHL-4, SUDHL-5, and HT (germinal center B-cell subtype). Each cell line exhibited a unique pattern of BCL-2 family protein expression, consistent with previous studies (Figure 1A; supplemental Figure 1A).34,35 Both BL cell lines were found to express BCL-W at levels detectable by western blotting; however, BCL-W protein could only be detected in 2 of the 4 DLBCL lines, namely HT and SUDHL-5 (Figure 1B). Transcriptional analysis for the mRNAs encoding BCL-W along with the other BCL-2 family prosurvival proteins were overall consistent with protein expression (Figure 1C; supplemental Figure 1B). Although it has been reported that BCL-W mRNA is significantly overexpressed in BL and DLBCL patient samples,26 we found higher transcriptional levels of BCL2L1 (BCL-X), MCL1, and BCL2 in 5 of the 6 lymphoma cell lines tested (supplemental Figure 1B). Of note, MCL1, which has been implicated in the sustained growth of MYC-driven lymphomas,36 was the most highly expressed of the prosurvival BCL-2 family members at the mRNA level in all 6 cell lines examined.
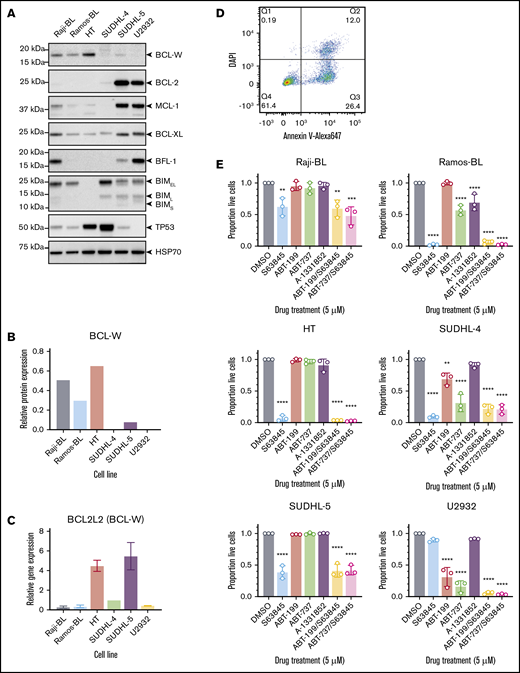
Characterization of select BL- and DLBCL-derived cell lines. (A) Western blotting for prosurvival proteins, proapoptotic BIM, and TP53 in human B-cell lymphoma lines. Probing for HSP70 was used as a loading control. Note that high TP53 protein levels in unstressed cells are indicative of tumor-driving mutations in the TP53 gene. (B) Quantification of BCL-W protein expression from representative western blotting experiments, normalized to the loading control, HSP70. (C) BCL-W gene expression measured by qRT-PCR. Data were normalized to HMBS housekeeping gene expression. (D) Flow cytometry gating strategy to determine cell viability. Live cells were Annexin V and DAPI double-negative. (E) Sensitivity profile of each cell line to 5 μM BH3-mimetic drugs: S63845 (MCL-1 inhibitor), ABT-199 (BCL-2 inhibitor), ABT-737 (BCL-2, BCL-XL, BCL-W inhibitor), and A-1331852 (BCL-XL inhibitor). Cell viability was measured as in panel D, and normalized to a DMSO-treated control sample. Error bars represent the SD for 3 independent experiments. Significance is indicated for comparisons to DMSO-treated control cells (1-way analysis of variance with Dunnett’s post hoc test, *P < .05, **P < .01, ***P < .001, ****P < .0001).
Characterization of select BL- and DLBCL-derived cell lines. (A) Western blotting for prosurvival proteins, proapoptotic BIM, and TP53 in human B-cell lymphoma lines. Probing for HSP70 was used as a loading control. Note that high TP53 protein levels in unstressed cells are indicative of tumor-driving mutations in the TP53 gene. (B) Quantification of BCL-W protein expression from representative western blotting experiments, normalized to the loading control, HSP70. (C) BCL-W gene expression measured by qRT-PCR. Data were normalized to HMBS housekeeping gene expression. (D) Flow cytometry gating strategy to determine cell viability. Live cells were Annexin V and DAPI double-negative. (E) Sensitivity profile of each cell line to 5 μM BH3-mimetic drugs: S63845 (MCL-1 inhibitor), ABT-199 (BCL-2 inhibitor), ABT-737 (BCL-2, BCL-XL, BCL-W inhibitor), and A-1331852 (BCL-XL inhibitor). Cell viability was measured as in panel D, and normalized to a DMSO-treated control sample. Error bars represent the SD for 3 independent experiments. Significance is indicated for comparisons to DMSO-treated control cells (1-way analysis of variance with Dunnett’s post hoc test, *P < .05, **P < .01, ***P < .001, ****P < .0001).
Testing the sensitivity of B-cell lymphoma cell lines to BH3-mimetic drugs
BH3-mimetic drugs have been developed that can individually target and inhibit BCL-2, BCL-XL, or MCL-1, as well as some that collectively target BCL-2, BCL-XL, and BCL-W. These compounds are useful to interrogate the dependency of cells on the different prosurvival BCL-2 proteins. The selected BL and DLBCL cell lines were treated with 5 μM each of S63845 (MCL-1 inhibitor), ABT-199/venetoclax (BCL-2 inhibitor), ABT-737 (BCL-2, BCL-XL, and BCL-W inhibitor), A-1331852 (BCL-XL inhibitor), or combinations thereof. After 24 hours, cell viability was determined by staining with Annexin V and DAPI, and flow cytometric analyses (Figure 1D-E). Ramos-BL, Raji-BL, HT, SUDHL-4, and SUDHL-5 cells all showed primary dependency on MCL-1, with only a minor further increase in apoptosis when S63845 was combined with either ABT-199 or ABT-737. U2932 cells showed dependency on BCL-2, yet the combination treatment with S63845 and ABT-199 was more effective than either single agent treatment alone (Student t tests, ABT-199 vs ABT-199/S63845: P < .05, S63845 vs ABT-199/S63845: P < .0001). This indicated that the survival of U2932 cells is safeguarded by both BCL-2 and MCL-1. Of particular interest, Ramos-BL, SUDHL-4, and U2932 cells were significantly more sensitive to ABT-737 than ABT-199 (Student t test, P < .05 for all), perhaps suggesting a potential minor role for BCL-W in the survival of these cell lines, although a reliance on BCL-XL is equally possible. Despite expressing BCL-W, as detected by western blotting, Raji-BL, HT, and SUDHL-5 cells were resistant to ABT-737 treatment, indicating no survival dependency on BCL-2, BCL-XL, or BCL-W, even collectively. For cell lines with a 50% inhibitory concentration (IC50) of ∼5 μM or less for a specific BH3-mimetic drug, a dose response curve was generated to calculate accurate IC50 values (supplemental Figure 1C).
Loss of BCL-W does not affect the survival of select human BL and DLBCL cell lines
Four of the characterized lymphoma cell lines, namely Ramos-BL, HT, SUDHL-4, and U2932, were selected for further analysis by CRISPR/CAS9 gene editing. SUDHL-5 cells were excluded because they rapidly silenced expression of the Cas9 transgene, and Raji-BL cells could not be infected with the Cas9 lentivirus. For 1 of the selected lines, Ramos-BL, the knockdown of BCL-W expression by shRNA has previously been shown to induce apoptosis in ∼50% of cells (Annexin V–positive) by 48 hours.25 To investigate the dependence of Ramos-BL cells and the selected DLBCL lines on BCL-W for sustained survival and growth, CRISPR/CAS9-mediated mutations were generated in BCL2L2 (henceforth referred to as BCL-W) via the nonhomologous end joining DNA repair pathway. Cell viability was monitored for 96 hours postinduction of CRISPR/CAS9 sgRNA expression by addition of doxycycline. In all cases, cells expressing CAS9 and a sgRNA targeting BCL-W (sgBCL-W) did not exhibit increased cell death compared with control cells expressing CAS9 and a nontargeting sgRNA (sgNT) to mouse Bcl2l11 (Bim) (Figure 2A). Disruption of the BCL-W gene was confirmed by sequencing after 5 days of sgRNA induction, which showed that for all cell lines containing BCL-W sgRNA, >50% of the total copies of the BCL-W–encoding gene possessed indels, primarily resulting in frameshifts (Figure 2B). Accordingly, substantial reductions in the expression of BCL-W protein in the Ramos-BL and HT cell lines carrying sgBCL-W were confirmed by western blot (Figure 2C) and qRT-PCR analyses (Figure 2D). The cell lines with reduced BCL-W expression were subsequently maintained in culture for >1 month, alongside parental and sgNT control cells. No effects on cell viability or cell growth were observed for any of the 4 lymphoma lines. Sequencing of the targeted region of BCL-W was repeated >1 month after sgRNA induction, with all 4 cell lines now showing almost complete mutation of the BCL-W gene (>87%) within the population. Thus, no selection against the silencing of BCL-W was apparent. To further confirm that BCL-W is dispensable for the survival of these cells, we generated single-cell clones of HT cells that had been transduced with either sgNT or sgBCL-W (supplemental Figure 2A-B). Several of the latter clones displayed no detectable BCL-W protein expression, as assessed by western blotting, and DNA sequencing revealed 100% loss of wild-type BCL-W gene sequences in all 6 HT clones tested, validating them as total BCL-W knockouts. As determined for the bulk populations, no cell death was observed upon loss of BCL-W in the HT-derived cell clones. These data indicate that BCL-W expression is not ubiquitously required for the sustained survival or growth of human BL or DLBCL cell lines, even for those that express detectable levels of BCL-W protein.
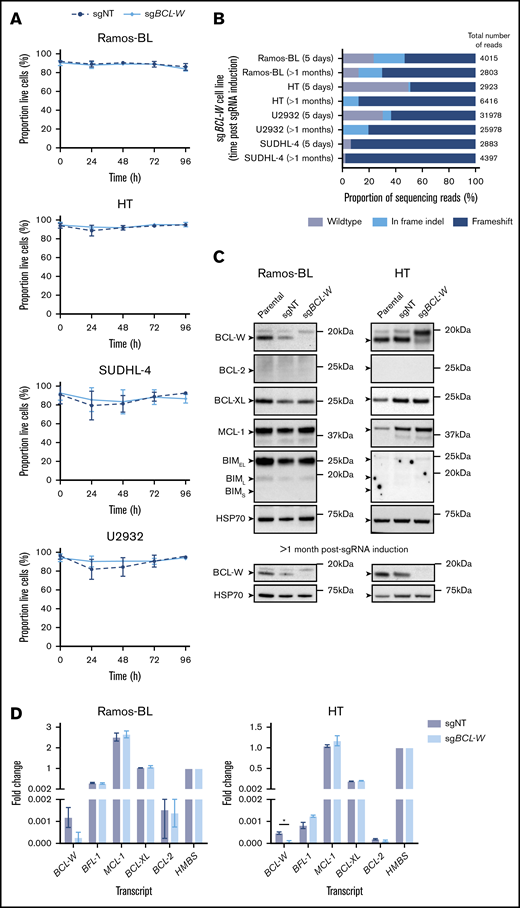
CRISPR/CAS9-mediated loss of BCL-W does not effect the survival of select BL and DLBCL cell lines. (A) Cell viability following doxycycline induction of nontargeting (sgNT) or BCL-W–targeting (sgBCL-W) sgRNAs over 96 hours. Cell viability was measured by the proportion of cells that were Annexin V and DAPI double negative by flow cytometry. Error bars represent the SD for 3 independent experiments. (B) Sequencing results for cell lines 5 days and >1 month postinduction of BCL-W sgRNA. Bars show the proportions of reads which were wild type, in frame indels, or frameshift in the BCL-W gene. The number of reads for each cell line is indicated. (C) Western blotting for prosurvival BCL-2 family members and proapoptotic BIM in sgBCL-W and control (sgNT) Ramos-BL and HT cells at 5 days and >1 month post-sgRNA induction. Probing for HSP70 was used as a loading control. (D) BCL-2 family gene expression in sgBCL-W and sgNT control Ramos-BL and HT cells as determined by qRT-PCR analysis. Data are normalized to HMBS housekeeping gene expression levels and shown relative to HMBS levels. Error bars represent the SD of 2 independent experiments. Significance is indicated (Student t test, *P < .05).
CRISPR/CAS9-mediated loss of BCL-W does not effect the survival of select BL and DLBCL cell lines. (A) Cell viability following doxycycline induction of nontargeting (sgNT) or BCL-W–targeting (sgBCL-W) sgRNAs over 96 hours. Cell viability was measured by the proportion of cells that were Annexin V and DAPI double negative by flow cytometry. Error bars represent the SD for 3 independent experiments. (B) Sequencing results for cell lines 5 days and >1 month postinduction of BCL-W sgRNA. Bars show the proportions of reads which were wild type, in frame indels, or frameshift in the BCL-W gene. The number of reads for each cell line is indicated. (C) Western blotting for prosurvival BCL-2 family members and proapoptotic BIM in sgBCL-W and control (sgNT) Ramos-BL and HT cells at 5 days and >1 month post-sgRNA induction. Probing for HSP70 was used as a loading control. (D) BCL-2 family gene expression in sgBCL-W and sgNT control Ramos-BL and HT cells as determined by qRT-PCR analysis. Data are normalized to HMBS housekeeping gene expression levels and shown relative to HMBS levels. Error bars represent the SD of 2 independent experiments. Significance is indicated (Student t test, *P < .05).
A potential explanation for the lack of cell death observed upon reduction or loss of BCL-W in DLBCLs and BLs is compensatory upregulation of an alternative prosurvival BCL-2 family member or downregulation of a proapoptotic BCL-2 family protein. To investigate this, bulk populations of gene-edited cells exhibiting loss of BCL-W and control cells with NT sgRNA were analyzed for prosurvival BCL-2, BCL-XL, MCL-1, and proapoptotic BIM protein expression after 5 days of sgRNA induction (Figure 2C). In all cases, no differences in expression of these proteins were observed between BCL-W–disrupted cells and control cells. Moreover, qRT-PCR analysis showed no significant differences in the mRNA levels of the nontargeted prosurvival BCL-2 family members in HT and Ramos-BL cells following disruption of BCL-W expression (Figure 2D).
Loss of BCL-W does not increase the susceptibility of human B-cell lymphoma-derived cell lines to BH3-mimetic drugs
Knockdown of BCL-W protein levels through the enforced expression of regulatory microRNAs has been reported to sensitize a range of human cancer-derived cell lines to chemotherapy, including non-small-cell and small-cell lung cancers, hepatocellular carcinoma, and clear cell renal cell carcinoma.37-40 BCL-W has also recently been identified in a CRISPR/CAS9 screen for factors rendering adenocarcinoma-derived cell lines resistant to combinations of BH3-mimetic drugs targeting BCL-2, MCL-1 or BCL-XL.28 Therefore, to determine whether loss of BCL-W sensitizes BL or DLBCL cells to BH3-mimetic drugs, we treated the bulk populations of sgNT (control) and sgBCL-W gene-edited Ramos-BL, HT, SUDHL-4, and U2932 cells with S63845 (MCL-1 inhibitor; note different doses were used depending upon the cell line), ABT-199 (BCL-2 inhibitor), ABT-737 (BCL-2/BCL-W/BCL-XL inhibitor), or A-1331852 (BCL-XL inhibitor). In all cases, the reduction of BCL-W protein did not significantly increase sensitivity of these particular lymphoma cell lines to any of the tested BH3-mimetic drugs following 24 hours of treatment (Figure 3). Furthermore, treatment of the HT BCL-W knockout clones gave results that were consistent with the analyses of the bulk cell populations, with no differences in sensitivity observed between BCL-W–deficient and control clones (supplemental Figure 2C-D). This was even the case when BCL-W-deficient clones were stressed by preculture in low-serum conditions (supplemental Figure 2E). These results indicate that BCL-W loss does not sensitize these B-cell lymphoma cells to BH3-mimetic drugs that target other BCL-2 prosurvival proteins.
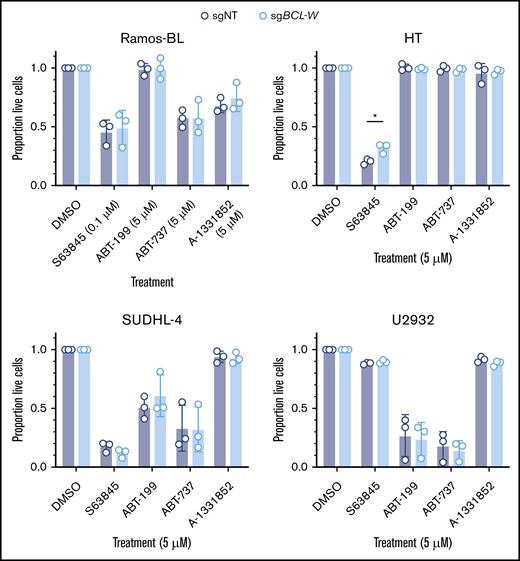
Loss of BCL-W does not sensitize BL or DLBCL cell lines to BH3-mimetic drugs. Viability of BCL-W–disrupted (sgBCL-W) and control (sgNT) cells following treatment with indicated dose of BH3-mimetic drugs for 24 hours: S63845 (MCL-1 inhibitor), ABT-199 (BCL-2 inhibitor), ABT-737 (BCL-2/BCL-XL/BCL-W inhibitor), and A-1331852 (BCL-XL inhibitor). Cell viability was measured by the proportion of cells that were Annexin V and DAPI double-negative by flow cytometry, and results were normalized to a DMSO-treated control sample. Loss of BCL-W did not sensitize any cell lines to any of the BH3-mimetic drugs tested (Student t test, P > .05 for all). Error bars represent the SD for 3 independent experiments. Significance is indicated (Student t test, *P < .05).
Loss of BCL-W does not sensitize BL or DLBCL cell lines to BH3-mimetic drugs. Viability of BCL-W–disrupted (sgBCL-W) and control (sgNT) cells following treatment with indicated dose of BH3-mimetic drugs for 24 hours: S63845 (MCL-1 inhibitor), ABT-199 (BCL-2 inhibitor), ABT-737 (BCL-2/BCL-XL/BCL-W inhibitor), and A-1331852 (BCL-XL inhibitor). Cell viability was measured by the proportion of cells that were Annexin V and DAPI double-negative by flow cytometry, and results were normalized to a DMSO-treated control sample. Loss of BCL-W did not sensitize any cell lines to any of the BH3-mimetic drugs tested (Student t test, P > .05 for all). Error bars represent the SD for 3 independent experiments. Significance is indicated (Student t test, *P < .05).
shRNA-mediated knockdown of BCL-W has no effect on survival or proliferation of select human BL- and DLBCL-derived cell lines
We sought to validate the finding that CRISPR/CAS9-mediated disruption of BCL-W expression had no effect on survival of the B-cell lymphoma cell lines using shRNA-mediated reduction of BCL-W expression. For these experiments, we selected the BL cell lines Raji-BL and Ramos-BL as shRNA-mediated knockdown of BCL-W has been shown to induce apoptosis in these cell lines,25 and the DLBCL cell line HT, which was found to express the highest levels of BCL-W of our cell line panel by western blotting. These cell lines were infected with lentiviral vectors containing doxycycline-inducible shRNAs against BCL-W (shBCL-W) or rat CD8 (nontargeting control; shNT), which have previously been published.31,32 BCL-W protein expression was examined by western blotting at 48 and 96 hours after shRNA induction, and was found to be efficiently knocked down in all 3 cell lines (Figure 4A; quantification of representative western blots at 96 hours compared to shNT control: Raji-BL: ∼88% knockdown, Ramos-BL: ∼70% knockdown, HT: ∼98% knockdown). Cell viability (Figure 4B) and proliferation (Figure 4C) were monitored during shRNA induction. Additionally, we measured the response of these cells to the BH3-mimetic drugs ABT-737 (BCL-2/BCL-XL/BCL-W inhibitor) and S63845 (MCL-1 inhibitor; note different doses were used depending on the cell line examined) following 72 hours of shRNA induction (Figure 4D). In all tests, no differences in cell viability, proliferation, or sensitivity to BH3-mimetic drugs were observed between BCL-W knockdown cells and cells expressing the nontargeting shRNA. Collectively, the results obtained from shRNA knockdown of BCL-W were consistent with our CRISPR/CAS9-mediated gene disruption studies, and suggest that anticancer therapies targeting BCL-W would be ineffective at killing the examined human BL- and DLBCL-derived cell lines.
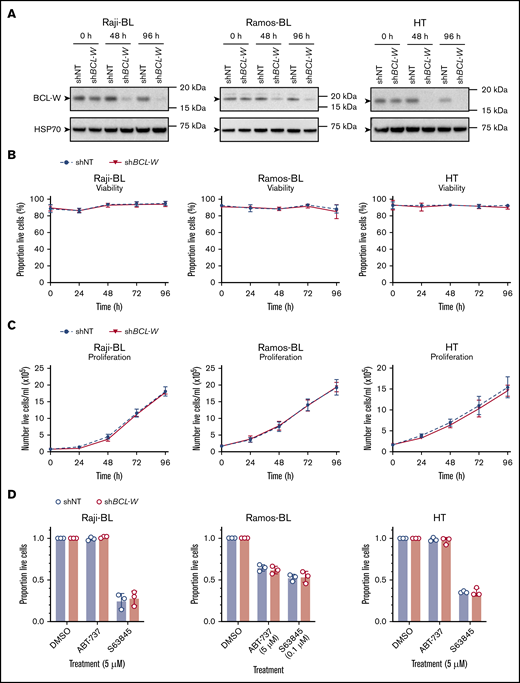
shRNA-mediated knockdown of BCL-W expression does not induce apoptosis of B-lymphoma cells and does not sensitize them to BH3-mimetic drugs targeting other prosurvival BCL-2 proteins. (A) shRNA knockdown of BCL-W was confirmed by western blotting in cell lines containing nontargeting control (shNT) and BCL-W–targeting (shBCL-W) shRNAs. Probing for HSP70 was used as a loading control. (B) Cell viability following doxycycline induction of shNT or shBCL-W expression over 96 hours. Cell viability was measured by the proportion of cells that were Annexin V and DAPI double-negative by flow cytometry. Error bars represent the SD for 3 independent experiments. (C) Cell proliferation following doxycycline induction of shNT or shBCL-W expression over 96 hours. Error bars represent the SD for 3 independent experiments. (D) Viability of BCL-W knockdown (shBCL-W) and control (shNT) cells following treatment with the indicated doses of BH3-mimetic drugs for 24 hours: S63845 (MCL-1 inhibitor), ABT-737 (BCL-2/BCL-XL/BCL-W inhibitor). Experiments were performed on cells following 72 hours of shRNA induction. Cell viability was measured by the proportion of cells that were Annexin V and DAPI double-negative by flow cytometry, and results were normalized to a DMSO-treated control sample. Knockdown of BCL-W did not sensitize any cell lines to ABT-737 or S63845 (Student t test, P > .05 for all). Error bars represent the SD for 3 independent experiments.
shRNA-mediated knockdown of BCL-W expression does not induce apoptosis of B-lymphoma cells and does not sensitize them to BH3-mimetic drugs targeting other prosurvival BCL-2 proteins. (A) shRNA knockdown of BCL-W was confirmed by western blotting in cell lines containing nontargeting control (shNT) and BCL-W–targeting (shBCL-W) shRNAs. Probing for HSP70 was used as a loading control. (B) Cell viability following doxycycline induction of shNT or shBCL-W expression over 96 hours. Cell viability was measured by the proportion of cells that were Annexin V and DAPI double-negative by flow cytometry. Error bars represent the SD for 3 independent experiments. (C) Cell proliferation following doxycycline induction of shNT or shBCL-W expression over 96 hours. Error bars represent the SD for 3 independent experiments. (D) Viability of BCL-W knockdown (shBCL-W) and control (shNT) cells following treatment with the indicated doses of BH3-mimetic drugs for 24 hours: S63845 (MCL-1 inhibitor), ABT-737 (BCL-2/BCL-XL/BCL-W inhibitor). Experiments were performed on cells following 72 hours of shRNA induction. Cell viability was measured by the proportion of cells that were Annexin V and DAPI double-negative by flow cytometry, and results were normalized to a DMSO-treated control sample. Knockdown of BCL-W did not sensitize any cell lines to ABT-737 or S63845 (Student t test, P > .05 for all). Error bars represent the SD for 3 independent experiments.
Discussion
The development of new BH3-mimetic drugs continues to be an area of great research focus due to the ongoing success of the BCL-2 inhibitor venetoclax for cancer therapy, and the promising preclinical evaluation of MCL-1 inhibitors, resulting in their entry into clinical trials for certain blood cancers in 2018.15,20,41 BCL-XL inhibitors have also been developed but progression in the clinic has been hampered by on-target toxicity to platelets.42 Attention is now shifting to address the therapeutic potential of targeting less well-studied BCL-2 family prosurvival members, such as BCL-W and BFL-1/A1. BCL-W is a particularly attractive anticancer target because loss of BCL-W has no effect on the normal healthy cells of mice, besides male sterility, suggesting it would be relatively well-tolerated by patients.2,3 We sought to examine the role of BCL-W in BLs and DLBCLs to ascertain whether BCL-W may be an effective drug target for the treatment of these malignancies, and ultimately whether BH3-mimetic drugs targeting BCL-W could have an effect in the clinic for the treatment of patients with these lymphomas.
We characterized BCL-2 family member expression in 6 human B-cell lymphoma–derived cell lines, detecting BCL-W protein by western blotting in only 4 of the lymphoma lines. Furthermore, qRT-PCR analysis showed that BCL-W mRNA was expressed at relatively low levels in these cell lines compared with the other prosurvival members MCL-1 and BCL-X. Interestingly, treatment of the parental cell lines with a range of BH3-mimetic drugs revealed that the MCL-1 inhibitor S63845 was the most effective at inducing cell death in 5 of the 6 lymphoma cell lines examined, suggesting a primary dependence on MCL-1 for sustained growth, as has been shown previously for BLs.36,43 To look directly at a role for BCL-W in sustained survival and growth of these lymphoma cell lines, we used CRISPR/CAS9 gene editing to successfully generate 3 DLBCL cell lines (HT, SUDHL-4, and U2932) and 1 BL cell line (Ramos-BL) exhibiting substantially reduced expression of BCL-W, as well as numerous single-cell–derived clones completely lacking BCL-W protein. In addition, we repeated these experiments in 2 of the previously listed cell lines, along with an additional cell line Raji-BL, using shRNA to knock down BCL-W expression. We found that loss of BCL-W expression by gene editing or shRNA did not lead to apoptosis in the human BL cell lines Ramos-BL and Raji-BL, nor did we observe any effect of BCL-W loss on the growth and survival of the 3 DLBCL-derived cell lines examined.
It has been shown that BH3-mimetic drugs can be more effective at killing certain types of cancers when used together in combinations44,45 ; therefore, we investigated this in the context of targeting BCL-W alongside other BCL-2 prosurvival proteins. We found that reduced or even complete loss of BCL-W expression did not increase the sensitivity of any of the lymphoma cell lines tested to treatment with BH3-mimetic drugs targeting the prosurvival proteins BCL-2, MCL-1, or BCL-XL individually, or targeting both BCL-XL and BCL-2 (and any remaining BCL-W) using ABT-737. Therefore, we anticipate therapeutic targeting of BCL-W alone would be ineffective at killing these B-cell lymphoma–derived cell lines and furthermore, it would not be expected to sensitize the tumor cells to BH3-mimetic drugs targeting other prosurvival BCL-2 family members.
In this study, we interrogated the expression of BCL-W at the RNA and protein levels using RNA knockdown, gene editing, and pharmacological approaches to disrupt BCL-W expression. From all experiments, our results indicate that BCL-W is dispensable for the sustained survival and growth of select human DLBCL- and BL-derived cell lines, despite its reported overexpression in patient samples and cell lines.25,26 Although the expression levels of prosurvival BCL-2 family members are generally thought to be good indicators of susceptibility to specific BH3-mimetic drugs,28,46 we found that this was not always the case. For example, U2932 cells predominantly express MCL1 by qRT-PCR, but show only negligible sensitivity to single-agent treatment with S63845. Thus, the high levels of BCL-W gene expression reported in some DLBCL patient samples may not necessarily indicate that these malignant cells are dependent on BCL-W for survival. Collectively, our results suggest that the development of new BH3-mimetic drugs that specifically target BCL-W will not be effective for all incidences of these B-cell lymphomas. However, it remains possible that BCL-W may play a role in the development of B-cell lymphomas and furthermore may be an effective therapeutic target in other human malignancies that we have not examined here.
Acknowledgments
The authors thank all members of the Molecular Genetics of Cancer Division at The Walter and Eliza Hall Institute (WEHI) for their support and advice; S. Monard and his team at the WEHI Flow Cytometry Unit; M. Herold and the MAGnetic Expansion Control (MAGEC) laboratory at WEHI for CRISPR/CAS9 and shRNA constructs; G. Lessene, C. White, and D. Huang for supplying aliquots of BH3-mimetic drugs; and R. Anderson and L. O’Reilly for antibodies. Next-generation sequencing was performed by Stephen Wilcox at the Ian Potter Centre for Genomics within the Walter and Eliza Hall Institute for Medical Research (Parkville, VIC, Australia).
This work was supported by funding from the Victorian Cancer Agency Fellowship (MCRF 17028) (G.L.K.), Cancer Council Victoria, grants-in-aid #1086157 and #1147328 (G.L.K.), the National Health and Medical Research Council (grant 1086291) (G.L.K.), Program grant 101671 (A.S.), Fellowship 1020363 (A.S.), Leukaemia Foundation Australia grant (G.L.K. and A.S.), Leukaemia and Lymphoma Society (grant 7001-13) (A.S.), the estate of Anthony (Toni) Redstone OAM, The Craig Perkins Cancer Research Foundation, and operational infrastructure grants through the Australian Government National Health and Medical Research Council Independent Research Institutes Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Support.
Authorship
Contribution: S.T.D., A.S., and G.L.K. conceived and designed the research; S.T.D., C.C., and L.T. performed the experiments; J.-n.G., P.L., A.C.D., and G.S.T. provided reagents; S.T.D., A.S., and G.L.K. analyzed data and interpreted results; S.T.D. drafted the manuscript; and A.S. and G.L.K. edited and revised the manuscript.
Conflict-of-interest disclosure: The Walter and Eliza Hall Institute receives milestone and royalty payments related to venetoclax, and employees (S.T.D., C.C., L.T., J.-n.G., A.S., and G.L.K.) may be eligible for benefits related to these payments. The laboratory of A.S. receives research funding from Servier. The remaining authors declare no competing financial interests.
Correspondence: Gemma L. Kelly, The Walter and Eliza Hall Institute, 1G Royal Parade, Parkville, VIC 3052, Australia; e-mail: gkelly@wehi.edu.au.

