Key Points
Outcomes of heavily pretreated high-risk CLL patients who undergo alloHCT after prior targeted therapy are excellent.
Immune reconstitution over time was faster for patients with prior targeted therapy.

Abstract
Allogeneic hematopoietic cell transplantation (alloHCT) can cure previously treated high-risk chronic lymphocytic leukemia (CLL) patients if they are suitable for transplant through the graft-versus-leukemia effect. However, since the emergence of targeted therapies, the role of alloHCT for high-risk CLL is less clear. To address this question, we evaluated 108 high-risk CLL patients who underwent alloHCT from 2010 to 2018. Thirty patients from the period of 2013 to 2018 received targeted therapy prior to alloHCT. The median age for the targeted therapy cohort was 60 years (range, 30-71 years), and 20% and 73% had complete and partial remission, respectively: 76% had del(17p), 46.2% had 5 or more cytogenetic abnormalities, and 78.9% were IGHV unmutated. The median number of prior therapies was 4 (range, 1-9). With a median follow-up time of 36 months (range, 10-72 months), the 3-year overall (OS) and progression-free survival (PFS) were 87% and 69%, respectively. The 3-year cumulative incidence of nonrelapse mortality and relapse was 7% and 24%, respectively. For the control cohort of 78 patients who underwent alloHCT from 2010 to 2014 and received only chemoimmunotherapy prior to transplant, the 3-year OS and PFS were 69% and 58%, respectively. Patients treated with targeted therapy prior to alloHCT had a significantly higher number of circulating T and B cells and a lower ratio of CD4 regulatory T cells to CD4 conventional T cells early after transplant. In summary, despite multiple high-risk features, the clinical outcome of CLL patients who receive targeted therapy prior to transplant is excellent and alloHCT should be offered while the disease is under control.
Introduction
Allogeneic hematopoietic cell transplantation (HCT; alloHCT) is an established treatment modality with curative potential in chronic lymphocytic leukemia (CLL). Prior to the advent of targeted therapies, alloHCT was recommended and widely accepted for high-risk CLL patients with relapsed/refractory disease after purine-analog–based chemotherapy and for patients with genetic features such as deletion of chromosome 17p [del(17p)] or TP53 mutation.1,2 However, with the widespread availability of targeted therapies such as ibrutinib, idelalisib, duvelisib, and venetoclax, the landscape of CLL treatment has changed markedly and the number of alloHCTs performed each year for CLL has substantially decreased.3,4 Despite this trend, however, the outcome after alloHCT has been steadily improving in safety and efficacy over time in both standard- and high-risk patients.5,6
Targeted therapies have proven efficacy but the prognosis of patients who receive targeted therapy after failure of chemoimmunotherapies (CITs) remains relatively poor if they have del(17p) and/or TP53 mutation.7 In addition, many high-risk patients discontinue targeted therapy due to the development of toxicity or eventually progress through all available agents.7-11 For those patients who have exhausted all therapies, few treatment options are available and alloHCT remains the only established potentially curative therapeutic modality. Nonetheless, identification of patients who will most benefit from alloHCT and the optimal timing of alloHCT have been less clear in the era of targeted therapy. In an effort to address these questions, the American Society of Blood and Marrow Transplantation published some recommendations for the use of alloHCT in CLL,12 and the European Research Initiative on CLL (ERIC) and the European Society for Blood and Marrow Transplantation (EBMT) proposed a transplant algorithm for patients with high-risk CLL treated with targeted therapies.7,13-16 However, only a few small studies have reported outcomes after alloHCT in patients who have received prior targeted therapies.16,17 CD19-targeted chimeric antigen receptor T cells (CD19 CAR-T) therapy has also been effective but the complete remission rate in relapsed/refractory CLL has been lower than in other B-cell maliganices.18-22 Although recent studies have been more encouraging, these studies are still quite small with relatively short follow-up periods.
In this study, we retrospectively investigated alloHCT outcome for high-risk CLL patients who failed CIT and received targeted therapies prior to alloHCT, and compared their baseline characteristics and clinical outcome with patients who only received CIT prior to alloHCT. Our goal was to ascertain whether patient characteristics have shifted, and whether transplant outcomes have been affected by the use of targeted therapy. We also examined T-, B-, and natural killer (NK)–cell recovery after alloHCT to investigate the impact of targeted therapy on immune reconstitution.
Materials and methods
Patient cohort
The Blood and Marrow Transplant data repository of the Dana-Farber Cancer Institute (DFCI) was queried to identify all patients with CLL, aged ≥18 years, who underwent alloHCT at the Dana-Farber/Brigham and Women’s Cancer Center between 1 January 2010 and 31 August 2018. After obtaining permission from the DFCI institutional review board and in accordance with the Declaration of Helsinki, retrospective chart review was performed to confirm the diagnosis of CLL: 108 patients were identified. Of these 108 patients, 30 received targeted therapy (defined as targeted small molecule therapy including Bruton tyrosine kinase, phosphatidylinositol 3 kinase, B-cell lymphoma 2, and investigational kinase inhibitors) mostly as salvage therapy prior to alloHCT; 78 only received CIT prior to alloHCT. With the emergence of targeted therapy for patients who failed CIT, targeted therapy was administered before considering alloHCT at our institute, with this transition starting in 2013. For patients who received prior targeted therapy, reasons for receiving alloHCT are listed in Table 1. The most common high-risk features included del(17p) (76%), IGHV-unmutated status (79%), ≥3 prior therapies (73%), and complex karyotype (≥5 abnormalities) (46%). All patients received reduced-intensity conditioning (RIC) HCT. The RIC regimens in this study included fludarabine with IV busulfan at doses of 3.2 mg/kg (Flu/Bu1; N = 55) or 6.4 mg/kg (Flu/Bu2; N = 48), fludarabine with melphalan (100-140 mg/m2) and low-dose total-body irradiation (TBI) 200 cGy (Flu/Mel/TBI) (N = 1), fludarabine with melphalan (100-140 mg/m2) and antithymocyte globulin (ATG; Flu/Mel/ATG) (N = 1), or fludarabine with cyclophosphamide (Cy) and low-dose TBI 200 cGy (Flu/Cy/TBI) (N = 3). Most of the patients received matched related or unrelated peripheral blood stem cell grafts (Table 2).
FISH
Retrospective chart review was performed to collect cytogenetic and fluorescence in situ hybridization (FISH) information. Patients with a standard metaphase karyotype analysis with at least 5 cells were considered evaluable, although the majority had the usual 20; patients were categorized as to normal karyotype or by the total number of abnormalities, with complex karyotype defined as 5 or more abnormalities. The choice of 5 abnormalities was based on our recent Center for International Blood and Marrow Transplant Research (CIBMTR) report23 as well as recent ERIC data.24 Patients with a FISH analysis were separately categorized as to presence or absence of del(13q), del(11q), del(17p), and trisomy 12.
Immunologic studies
The full description of immunologic studies is provided in supplemental Material.
Statistical analysis
Baseline characteristics were reported descriptively and compared using the Fisher's exact test, the χ2 test, or the Wilcoxon rank-sum test as appropriate. The primary end point was overall survival (OS) and other end points of interest included progression-free survival (PFS), relapse, and nonrelapse mortality (NRM). All time-to-event end points were measured from stem cell infusion to death (OS, NRM) or death or relapse (PFS, relapse). OS and PFS were estimated using the Kaplan-Meier method; the log-rank test was used for comparisons of Kaplan-Meier estimates. Cumulative incidence curves for NRM and relapse were constructed in the competing-risks framework considering relapse and NRM as a competing event, respectively. The difference between cumulative incidences in the presence of a competing risk was tested using the Gray method.25 Univariable and multivariable Cox regression analysis was performed to examine factors that are associated with OS and PFS. For the multivariable model, high-risk features or factors that were associated with P < .1 from univariable models were included. Risk factors considered in regression analysis included age, patient sex, patient and donor sex combination, graft source, donor HLA type, RIC intensity, sirolimus use as graft-versus-host disease (GVHD) prophylaxis, disease status at alloHCT, patient-donor cytomegalovirus serostatus, HCT-comorbidity score,26 Richter transformation, number of prior therapies, white blood cell count, percentage of bone marrow involvement, lactate dehydrogenase (LDH), immunoglobulin heavy chain variable region (IGHV) mutation status, FISH, complex karyotype, and time from first CLL therapy to alloHCT. Year of transplant correlated highly with type of prior therapy (r = 0.78; P < .0001) and thus was not considered in the Cox model to avoid a collinearity issue23 as this was represented by the type of prior therapy. For the targeted therapy cohort, duration of targeted therapies was also examined in univariable analysis. Prior to modeling, linearity assumption for continuous variables, the proportional hazards assumption, and significance of 2-way interaction terms were examined. For selection of a best model, Akaike information criteria were used.27 Firth correction was applied to reduce the bias due to the limited number of events.28,29 For comparison of laboratory parameters, the Wilcoxon rank-sum test was used. Multiplicity was not considered. All P values were 2-sided and the significance level was set to .05. All analyses were performed using SAS 9.4 (SAS Institute Inc, Cary, NC), and R version 3.6.1 (the CRAN project; www.cran.r-project.org).
Results
Patients
The baseline characteristics of all patients are summarized in Table 2. Thirty patients received targeted therapies prior and subsequently underwent RIC alloHCT from 2013 to 2018 (targeted therapy cohort). Twenty-eight of 30 patients had a prior history of disease progression and/or refractoriness while receiving CIT prior to receiving targeted therapy. Two patients who had received a single prior therapy, ibrutinib, without a history of receiving CIT before alloHCT underwent alloHCT due to high-risk features of del(17p) and IGHV unmutated (Figure 1). Seventy-three percent of patients were in partial remission at the time of alloHCT and 6 patients (20%) were in complete remission. The median number of total prior therapies (combined CIT and targeted) was 4 (range, 1-9). The median duration of targeted therapy was 10.5 months (range, 2.8-53.1) and the median time from first CLL therapy to alloHCT was 39.5 months (range, 6.7-173.3). Nineteen (63%) had received ibrutinib only, and 3 had received venetoclax only (10%), mostly after failure of CIT; 8 (27%) had received multiple targeted therapies prior to alloHCT (Figure 1; Table 2). Patients who received multiple targeted therapies prior to alloHCT were more recently transplanted. For a control cohort, 78 patients who underwent alloHCT from 2010 to 2013 received CIT prior to alloHCT. The median number of prior CIT was 3 (range, 1-10). Baseline characteristics did not differ significantly between the 2 cohorts except for the RIC regimen, white blood cell count, year of transplant, and frequency of del(17p). More patients in the targeted therapy cohort had del(17p) and received Flu/Bu2 as the institute gradually moved its practice from Flu/Bu1 to Flu/Bu2 in recent years. In addition, day 30 total cell chimerism was similar between the 2 cohorts: median chimerism was 97% (range, 51-100) vs 95% (range, 59-100) for the targeted therapy and CIT, respectively (P = .72).
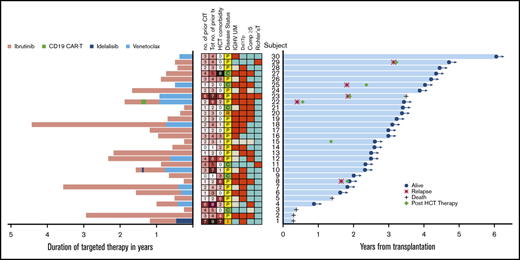
High-risk features, duration of targeted therapy prior to allogeneic HCT, and time to event. (A) Duration of each targeted therapy is denoted in pink (ibrutinib), green (CD19 CAR-T), dark blue (idelalisib), and light blue (venetoclax). Of note, time between targeted therapies is not reflected in the duration of targeted therapy. In the center, boxes in red indicate subjects with IGHV unmutated (UM), del(17p), complex abnormalities (≥5), or Richter transformation. Boxes in blue indicate no mutation or no Richter transformation. Boxes in gray indicate missing information. Tot no. of prior tx denotes the total number of targeted and chemoimmunotherapies received prior to alloHCT. No. of CIT denotes the total number of chemoimmunotherapies prior to targeted therapy. Disease status denotes complete remission (C), partial remission (P), relapse (R), or induction failure (I) at alloHCT. (B) The blue horizontal bars indicate time to death or last seen alive with the indication of relapse and post-HCT therapy.
High-risk features, duration of targeted therapy prior to allogeneic HCT, and time to event. (A) Duration of each targeted therapy is denoted in pink (ibrutinib), green (CD19 CAR-T), dark blue (idelalisib), and light blue (venetoclax). Of note, time between targeted therapies is not reflected in the duration of targeted therapy. In the center, boxes in red indicate subjects with IGHV unmutated (UM), del(17p), complex abnormalities (≥5), or Richter transformation. Boxes in blue indicate no mutation or no Richter transformation. Boxes in gray indicate missing information. Tot no. of prior tx denotes the total number of targeted and chemoimmunotherapies received prior to alloHCT. No. of CIT denotes the total number of chemoimmunotherapies prior to targeted therapy. Disease status denotes complete remission (C), partial remission (P), relapse (R), or induction failure (I) at alloHCT. (B) The blue horizontal bars indicate time to death or last seen alive with the indication of relapse and post-HCT therapy.
Clinical outcome: targeted therapy cohort
The duration of prior targeted therapies, events of relapse, post-HCT therapy, and duration of OS for all 30 patients are depicted in Figure 1. The figure also depicts the high-risk features: del(17p), complex karyotype (defined as ≥5 abnormalities22,23 ), Richter transformation and IGHV-unmutated status, total number of prior targeted and CIT therapies, HCT-comorbidity score, and disease status at alloHCT; no association between the OS time and these high-risk features (except 1 patient [subject 1]) was apparent. Two long-term survivors (subjects 22 and 23) had 6 and 7 prior therapies and relapsed after alloHCT; were rescued by post-HCT therapy. Subject 22 received 6 prior therapies including ibrutinib, CD19 CAR-T, second ibrutinib, and venetoclax prior to HCT, with venetoclax for early post-HCT relapse, and is still alive at 41 months after alloHCT. Subject 23 had Richter transformation; was treated with 7 prior therapies including venetoclax prior to HCT; received ibrutinib and rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) post-HCT relapse; and died of infection at 42 months after HCT.
Three patients had Richter transformation (subjects 11, 23, 29). Two had partial remission (PR) and 1 had complete remission (CR) at the time of alloHCT. Subject 11 had 5 prior therapies including ibrutinib and R-CHOP and is still alive and relapse-free at 28 months post-HCT. Subject 29 had 4 prior therapies and received R-CHOP and donor lymphocyte infusion (DLI) for post-HCT relapse. This patient is still alive at 56 months after alloHCT.
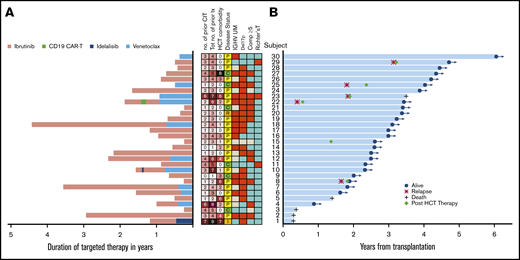
For the entire targeted cohort, the median follow-up among survivors was 36 months (range, 10.3-72.3 months) and the median OS and PFS were not reached (Figure 2). Overall, only 5 of 30 patients died and 1 patient relapsed prior to death. Four additional patients relapsed without death. The 3-year OS was 87% (95% confidence interval [CI], 68%, 95%), 3-year PFS was 72% (95% CI, 52%, 85%), 3-year cumulative incidence of NRM was 7% (95% CI, 1%, 19%), and 3-year cumulative incidence of relapse was 21% (95% CI, 8%, 38%) (Table 3; Figure 2A-B).
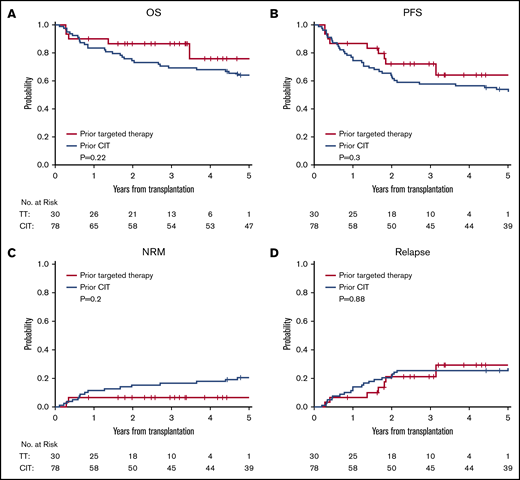
Kaplan-Meier and cumulative incidence curves. OS (A), PFS (B), cumulative incidence of NRM (C), and cumulative incidence of relapse (D) according to the type of prior therapy before alloHCT.
Kaplan-Meier and cumulative incidence curves. OS (A), PFS (B), cumulative incidence of NRM (C), and cumulative incidence of relapse (D) according to the type of prior therapy before alloHCT.
To identify subsets of patients who benefit the most or the least from alloHCT, we performed univariable Cox regression analysis for OS for the patient and transplant characteristics listed in Table 2. The only factor that was associated with increased risk of death was the HCT-comorbidity score (hazard ratio [HR], 1.4; 95% CI, 1.03, 1.91; P = .032). Due to the small number of events, multivariable analysis was not possible. HCT-comorbidity score as a continuous variable was also the only significant factor (HR, 1.26; 95% CI, 1.02, 1.57; P = .036) for PFS. No effect of disease status was seen when comparing CR and PR (supplemental Figure 3); relapse and induction failure at HCT could not be properly assessed, with only 1 patient each. The 1 patient who was relapsed at HCT remains alive without disease progression 40.2 months later, and the patient refractory to idelalisib at HCT died at day 100 (subject 1). Neither FISH abnormalities nor complex karyotype was significant in predicting PFS or OS. Furthermore, exposure to multiple targeted therapies prior to alloHCT, type of targeted therapy, duration of targeted therapy, or time from first CLL therapy to alloHCT did not affect OS or PFS.
Comparison between the targeted and chemoimmunotherapy cohorts
We then compared alloHCT outcome between the targeted therapy cohort and the control cohort. The outcome for each cohort and both cohorts combined (N = 108) is presented in Table 3 and Figure 2. For both cohorts combined, the 3-year OS and PFS were 73% and 61%, respectively; 3-year cumulative incidence of NRM and relapse was 14% and 24%, respectively; 6-month grade III-IV acute GVHD was 9%, and the 1-year cumulative incidence of chronic GVHD was 48%. Although most outcomes trended better in the targeted therapy cohort, none were statistically superior to the control cohort in this univariable analysis: 3-year OS, 87% vs 69%, respectively (P = .22); 3-year PFS, 72% vs 58% (P = .3); cumulative incidence of NRM at 3 years, 7% vs 17% (P = .2); and 3-year relapse, 21% vs 26% (P = .88). No difference was seen in acute or chronic GVHD either; 6-month grade III-IV acute GVHD was 13% vs 7.7% (P = .5) and 1-year chronic GVHD 57% vs 45% (P = .47).
In a multivariable Cox model for both cohorts combined, the HR for the targeted therapy cohort relative to the control cohort was 0.35 (95% CI, 0.13, 0.97; P = .043) for OS and 0.47 (95% CI, 0.22, 1.03; P = .06) for PFS. Other factors that were significant in OS were nonbusulfan-based conditioning regimen (HR, 5.1; 95% CI, 1.14, 22.8; P = .033) and Richter transformation (HR, 3.62; 95% CI, 1.01, 12.9; P = .048). Factors that were associated with PFS included male recipient with female donor (HR, 2.28; P = .02), HCT-comorbidity score (HR, 1.17; P = .036), nonbusulfan-based conditioning regimen (HR, 4.22; P = .045), and Richter transformation (HR, 3.97; P = .005). Busulfan dose (Flu/Bu1 vs Flu/Bu2) was not significantly associated with OS and PFS (supplemental Table 2).
Post-HCT therapy
Of 108 patients, 23 patients received post-HCT therapy for posttransplant relapse including 1 impending relapse: 6 (20%) in the targeted therapy cohort and 17 (22%) in the control cohort. The post-HCT therapies included ibrutinib alone (N = 9), ibrutinib with other therapies (N = 3), alemtuzumab plus or minus high-dose methylprednisolone (N = 2), radiation (N = 2), DLI plus CIT (N = 1), and rituximab plus or minus other (N = 2). Two of 6 patients who received post-HCT therapy in the targeted therapy cohort had Richter transformation (subjects 23, 29); 1 received ibrutinib plus R-CHOP and the other received R-CHOP and then DLI. Detailed information on post-HCT therapy is listed in supplemental Table 3. For these 23 patients, the median OS from the initiation of post-HCT therapy has not been reached and the 3-year OS was 61% (95% CI, 37%, 79%). In the targeted cohort, 5 patients who received post-HCT therapy are still alive and 1 died of disease after 19.3 months of the post-HCT therapy.
Immunologic correlates
Immunophenotypic analysis by flow cytometry was performed prospectively at defined intervals posttransplant using whole blood. Compared with patients with prior CIT, patients with prior targeted therapy had a higher lymphocyte count (P = .03), circulating CD3+ cells (P = .006), CD4+ cells (P = .002), and normal B cells (CD19+CD5−) (P = .001) at 1 month post-alloHCT (Figure 3A). Patients with prior targeted therapy had a higher fraction of CD4 conventional T cells (CD4Tcons) relative to CD4 regulatory T cells (CD4+Tregs), which led to a significantly lower CD4Treg/CD4Tcon ratio during the first 6 months after alloHCT (P = .003, .02, .004, .016 at 1, 2, 3, 6 months post-alloHCT, respectively; Figure 3B). In addition, within the CD4Tregs, patients with targeted therapy had a smaller fraction of central memory (CM) cells at 3 and 18 months (P = .025 and .04, respectively) and naive cells at 1, 2, and 6 months (P = .008, .026, .03, respectively), but a higher fraction of effector memory (EM) cells at 3 and 6 months (P = .01 and .02, respectively) and terminally differentiated (TD) cells (P = .018 at 2 months, P < .001 at 3, 6, 9, 12, 18 months). Similarly, within CD4Tcons, patients with targeted therapy had a smaller fraction of CM cells at 1, 2, 6, and 9 months (P = .003, .02, .006, .03, respectively) and naive cells at 1 month (P = .03), but a higher fraction of EM cells at 3 and 6 months (P = .009 and .03, respectively) and TD cells at all time points (P < .01 at 1, 2, 3, 6, 9, 12, 18 months and P = .048 at 24 months; Figure 3C). Furthermore, B-cell reconstitution was significantly faster in the targeted therapy cohort compared with the control cohort (P = .001, .03, .003, .005, .05 at 1, 2, 3, 6, 9 months; Figure 3D). The majority of B cells were antigen-naive B cells (CD19+CD5−27−) and the number of circulating antigen-naive B cells was significantly higher in the targeted therapy compared with the control cohort at 1, 3, and 6 months (P = .006, .06, .03, .009 at 1, 2, 3, 6 months, respectively). To rule out the impact of conditioning intensity on immune reconstitution, the analysis was repeated for patients who received Flu/Bu2 only and the result was consistent (supplemental Figure 4).
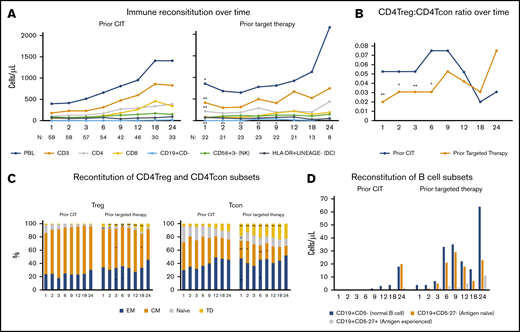
Assessment of immunologic correlative studies. (A) Time course of major cell populations at baseline and through 2 years of alloHCT, including lymphocyte counts (PBL), total T cells (CD3+), CD4+ and CD8+ T cells, B cells (CD19+CD5−), NK cells (CD56+CD3−), and dendritic cells (HLA-DR+LINEAGE−). Each dot represents median value of absolute counts. (B) CD4Treg/CD4Tcon ratio over time. (C) CD4Treg and CD4Tcon median percentage of effector memory (EM), central memory (CM), naive and terminally differentiated (TD) cells. (D) Absolute counts of B cells (CD19+CD5−), antigen-naive B cells (CD19+CD5−27−), and antigen experienced B cells (CD19+CD5−27+). Each dot represents median value. *P < .05 and **P < .01 compared with the prior chemoimmunotherapy.
Assessment of immunologic correlative studies. (A) Time course of major cell populations at baseline and through 2 years of alloHCT, including lymphocyte counts (PBL), total T cells (CD3+), CD4+ and CD8+ T cells, B cells (CD19+CD5−), NK cells (CD56+CD3−), and dendritic cells (HLA-DR+LINEAGE−). Each dot represents median value of absolute counts. (B) CD4Treg/CD4Tcon ratio over time. (C) CD4Treg and CD4Tcon median percentage of effector memory (EM), central memory (CM), naive and terminally differentiated (TD) cells. (D) Absolute counts of B cells (CD19+CD5−), antigen-naive B cells (CD19+CD5−27−), and antigen experienced B cells (CD19+CD5−27+). Each dot represents median value. *P < .05 and **P < .01 compared with the prior chemoimmunotherapy.
Discussion
Allogeneic transplant outcomes have been continuously and steadily improving over the past 40 years due to advances in donor source, conditioning regimen, high-resolution HLA typing, GVHD prophylaxis, and supportive care.5,6 Consistent with this observation, we report a very favorable outcome for previously treated high-risk CLL patients who underwent RIC HCT in recent years. The outcome for patients who received targeted therapy after failure of CIT prior to alloHCT was excellent (3-year OS and PFS, 87% and 72%, respectively) despite the fact that these patients had already failed multiple targeted and/or CIT and transplant was offered in most cases as a last resort. With use of RIC, the NRM rate (7%) was low without overt increase of relapse (21%). This outcome reported in the current study compares favorably to previously published outcomes in high-risk CLL. To explore the feasibility of the ERIC/EBMT transplant algorithm, in Hoffman et al,17 a small group of patients was categorized into a low- and a high-transplant-risk group; the 2-year OS and PFS were 95% and 68%, respectively, in the low-transplant-risk group and 65% and 56%, respectively, in the high-transplant-risk group.17 The low-transplant-risk group was defined as patients with age ≤65 years, del(17p), and/or TP53 abnormalities, failed CIT but responding to first targeted therapy, no comorbidity, and well-matched donor. According to this definition, 5 patients in our targeted cohort fell into the low-transplant-risk group and the 3-year OS and PFS were 100% and 80%, respectively; the 3-year OS and PFS in the high-transplant-risk group were 84% and 71%, respectively. In another small study for pretransplant ibrutinib-sensitive high-risk patients, Dreger et al reported 1-year PFS of 65%.16 There have also been a few CD19-directed CAR–T-cell studies in relapsed/refractory CLL. In Porter et al,18 14 patients with relapsed/refractory CLL were treated with CD19-CAR-T and the 18-month OS and PFS was 71% and 28.6%, respectively. In the subsequent long-term follow-up study with more patients enrolled,19 Frey et al reported 3-year OS and PFS of 62% and 26%, respectively. In a pilot study to assess concurrent administration of ibrutinib with CAR-T in relapsed/refractory CLL (N = 18), Gauthier et al reported 1-year OS and PFS of 64% and 38%, respectively, for concurrent ibrutinib plus CAR-T and 61% and 50%, respectively, for CAR-T alone.20,21 In anti-CD19 CAR–NK-cell therapy, 5 patients with relapsed/refractory CLL received CAR–NK-cell therapy.22 Of these 5, 4 patients received postremission therapy between 1 and 9 months of the infusion, thus making it impossible to determine the duration of response. One patient did not achieve remission and received alloHCT at 6 months of infusion and remained disease-free.
Our data suggest that, for patients treated with targeted therapy, high-risk features such as del(17p) or complex karyotype (≥5 abnormalities) or depth of remission at the time of alloHCT (CR vs PR) did not significantly impact alloHCT outcome, although confirmation of this observation warrants a larger study. On the other hand, HCT-comorbidity score showed a weak correlation with the outcome as 1 patient with comorbidity score 8 is still alive and another patient with comorbidity score 6 survived for 42 months, indicating that HCT can still benefit patients with a high comorbidity score if disease is reasonably controlled, although in general a lower HCT-comorbidity score is more optimal.
When we examined whether there was a shift in baseline characteristics in recent years, we noted that patients in both cohorts were heavily pretreated with high-risk CLL and the targeted cohort had even more patients with del(17p). This is not surprising, as targeted therapies can result in long-term remission in lower-risk patients. In contrast, the prognosis of high-risk patients [eg, del(17p)] treated with a first targeted therapy after CIT failure remains poor, with a higher likelihood of transforming to Richter syndrome.7,8 For this reason, along with our prior experience and other reports,7,23,30 we believe that disease control at the time of alloHCT is of paramount importance for the success of alloHCT. Thus, until prospectively proven otherwise, our current practice is to offer alloHCT to previously treated CLL patients with del(17p) or ≥5 cytogenetic abnormalities on karyotype, or Richter transformation, while their disease is reasonably controlled, typically on a second targeted therapy, as these patients are likely to relapse relatively early on targeted therapy.7,31 In regard to age, due to the favorable tolerability of RIC, we offer alloHCT to patients up to 75 years of age.
Our immune correlative analyses provide some insights into the effect of targeted therapy on immune reconstitution. We found that patients who received prior targeted therapy had significantly higher circulating T-cell counts early after alloHCT and a significantly lower CD4Treg/CD4Tcon ratio up to 6 months after alloHCT compared with those who received prior CIT. This result was consistent when we limited to patients receiving Flu/Bu2 only. In our previous phase 3 study of administering ATG prior to stem cell infusion, we found that all T cells were substantially suppressed early after alloHCT in the ATG arm compared with the placebo arm.32 This result suggests that the prior regimen profoundly affects the post-HCT immune reconstitution. We also found that B-cell reconstitution is faster in the targeted therapy cohort compared with the control cohort. This is possibly due to the relatively nonimmunosuppressive nature of targeted therapy compared with CIT. Although many studies including our own33-36 have reported a positive correlation between lymphocyte reconstitution and clinical outcome, whether this robust lymphocyte reconstitution is associated with fewer infections and/or a stronger graft-versus-leukemia effect warrants further investigation.
Our study is subject to all the inherent limitations of a single-center retrospective review with a relatively small number of patients who underwent alloHCT in the era of targeted therapy. Because all patients who underwent alloHCT in recent years received prior targeted therapy, whether the excellent outcome seen in the targeted cohort is due to advances in alloHCT5,6 (including a more intensive conditioning regimen) or due to the prior targeted therapy or both is unclear. To gain some insights into this question, we examined OS for all of our patients with lymphoid malignancies who received RIC HCT from 2010 and 2018 (N = 579) at our institute and found that the improvement was incremental: the 2-year OS 67% from 2010 to 2012, 74% from 2013 to 2015, and 79% after 2016. Therefore, it is likely that both improvements in alloHCT management in recent years and rapid immune reconstitution contribute to the excellent outcome seen in the targeted therapy cohort.
In summary, in the midst of the emergence of targeted therapies, alloHCT has made steady progress in improving outcomes. Disease clearance by graft-versus-leukemia after alloHCT is known to be durable and effective across all genetically defined high-risk subsets14 and this is again supported by our study. As many high-risk patients who are treated with targeted therapy will eventually progress or discontinue the therapy due to toxicity, an integrative approach of transplant and targeted therapy, that is, inducing remission in high-risk patients with targeted therapy, offering alloHCT during remission, and potentially reinstating targeted therapy for consolidation post-HCT or post-HCT relapse, might enhance the clinical outcome of these patients.16,23
All reasonable requests for raw and analyzed data that are not included in this manuscript or online content will be promptly reviewed by the senior authors to determine whether the request is subject to any intellectual property or confidentiality obligations. Patient-related data may be subject to patient-confidentiality restrictions. Any data and materials that can be shared will be released via a material transfer agreement. E-mail the corresponding author, Haesook T. Kim, at htkimc@jimmy.harvard.edu.
Acknowledgments
The authors thank the patients and their families, the research coordinators, research nurses, and advanced practice providers.
This work was supported by research funding from the National Institutes of Health, National Cancer Institute (P01CA229092) and the Ted and Eileen Pasquarello Tissue Bank in Hematologic Malignancies. C.J.W. is a Scholar of the Leukemia & Lymphoma Society. J.R.B. was supported by the Susan and Gary Rosenbach Fund for Lymphoma Research, the Melton Family Fund for CLL Research, and National Institutes of Health, National Cancer Institute grant R01 CA213442-01A1.
Authorship
Contribution: H.T.K. conceived and designed the study, performed statistical analysis, interpreted the data, and wrote the manuscript; C.J.S. and J.R.B. compiled the outcome data, provided FISH data, and annotated the cytogenetic data; S.C.R., C.R., and J.R. provided the flow cytometry data; J.R.B. edited the manuscript; and all authors contributed to the manuscript review and approved the final version for submission.
Conflict-of-interest disclosure: J.R.B. has served as a consultant for AbbVie, Acerta, AstraZeneca, Beigene, Catapult, Dynamo Therapeutics, Juno/Celgene, Kite, MEI Pharma, Nextcea, Novartis, Octapharma, Pfizer, Sunesis, TG Therapeutics, and Verastem; received honoraria from Janssen and Teva; received research funding from Gilead, Loxo, Sun, and Verastem; and served on data safety monitoring committees for Morphosys and Invectys, outside of the submitted work. C.J.W. is a cofounder and member of the scientific advisory board of Neon Therapeutics; and receives research funding from Pharmacyclics, Inc, outside of the submitted work. J.R. has received research funding from Amgen, Equillium, and Kite Pharma; and served as a consultant for Aleta Biotherapeutics, Avrobio, Celgene, Falcon Therapeutics, LifeVault Bio, Rheos Medicines, Talaris Therapeutics, and TScan Therapeutics, outside of the submitted work. J.K. has served as a consultant for Amgen, Equillium, Cugene, and Moderna; served on an advisory board for Therakos; and received funding from Bristol Myers Squibb (BMS), Miltenyi, Clinigen, and Regeneron, outside of the submitted work. P.A. has served as a consultant for Merck, BMS, Pfizer, Affimed, Adaptive, Infinity, ADC Therapeutics, Celgene, Morphosys, Daiichi Sankyo, Miltenyi, Tessa, GenMab, C4, and Enterome; received research funding from Merck, BMS, Affimed, Adaptive, Roche, Tensha, Otsuka, Sigma Tau, Genentech, and IGM; and received honoraria from Merck and BMS, outside of the submitted work. R.J.S. has served as a consultant for Gilead, Rheos Therapeutics, Jazz, Cugene, Mana Therapeutics, VOR, and Novartis; served on data safety monitoring committees for Juno; and served on the board of directors for Kiadis and Be the Match/National Marrow Donor Program (NMDP), outside of the submitted work. H.T.K. has served as a consultant for Miltenyi, outside of the submitted work. The remaining authors declare no competing financial interests.
The current affiliation for E.P.A. is Department of Medicine, Duke University School of Medicine, Durham, NC.
Correspondence: Haesook T. Kim, Department of Data Sciences, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA 02215; e-mail: htkimc@jimmy.harvard.edu.