Key Points
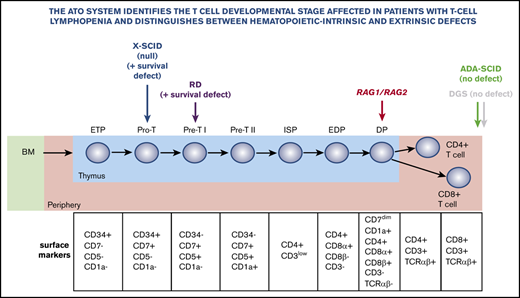
The ATO system allows precise definition of developmental blocks in patients with gene defects that cause T-cell lymphopenia.
The ATO system can help distinguish between hematopoietic autonomous and extra-hematopoietic defects that cause T-cell lymphopenia.

Abstract
The study of early T-cell development in humans is challenging because of limited availability of thymic samples and the limitations of in vitro T-cell differentiation assays. We used an artificial thymic organoid (ATO) platform generated by aggregating a DLL4-expressing stromal cell line (MS5-hDLL4) with CD34+ cells isolated from bone marrow or mobilized peripheral blood to study T-cell development from CD34+ cells of patients carrying hematopoietic intrinsic or thymic defects that cause T-cell lymphopenia. We found that AK2 deficiency is associated with decreased cell viability and an early block in T-cell development. We observed a similar defect in a patient carrying a null IL2RG mutation. In contrast, CD34+ cells from a patient carrying a missense IL2RG mutation reached full T-cell maturation, although cell numbers were significantly lower than in controls. CD34+ cells from patients carrying RAG mutations were able to differentiate to CD4+CD8+ cells, but not to CD3+TCRαβ+ cells. Finally, normal T-cell differentiation was observed in a patient with complete DiGeorge syndrome, consistent with the extra-hematopoietic nature of the defect. The ATO system may help determine whether T-cell deficiency reflects hematopoietic or thymic intrinsic abnormalities and define the exact stage at which T-cell differentiation is blocked.
Introduction
Limited access to thymic samples and the relative inefficiency of in vitro T-cell development methods have hampered precise definition of the developmental blocks that characterize different forms of severe combined immune deficiency (SCID) in humans. A serum-free 3D artificial thymic organoid (ATO) system has recently been shown to support human T-cell differentiation efficiently and reproducibly in vitro from hematopoietic stem cells. It has advantages over previously published protocols for its technical simplicity, reliability, and efficient production of cells.1 Here, we used the ATO system to define developmental blocks in patients with genetic defects that cause T-cell lymphopenia of variable severity and to assess the power of the system to distinguish between hematopoietic autonomous and extra-hematopoietic causes of T-cell lymphopenia.
Methods
Isolation of human CD34+CD3– hematopoietic stem and progenitor cells
CD34+ cells purified from granulocyte colony-stimulating factor/plerixafor-mobilized peripheral blood (MPB) samples were obtained from adult normal donors (NDs) who were undergoing apheresis for allogeneic stem cell transplant donation at the National Institutes of Health (NIH) or from patients undergoing autologous stem cell transplantation. Bone marrow (BM) aspirates were obtained from patients admitted to the NIH Clinical Center or sent in from other centers in the United States. Their blood was enriched for mononuclear cells by gradient centrifugation using Ficoll-Paque (GE Healthcare Life Sciences, Pittsburgh, PA) before cryopreservation or flow cytometry sorting. The study was conducted according to protocols 94-I-0073, 18-I-0041, and 18-I-N128 and was approved by the NIH Institutional Review Board. Informed consent was provided by patients and their parents.
ATO generation and culture
The ATOs were generated by aggregating a DLL4-expressing stromal cell line (MS5-hDLL4) with CD34+ cells isolated from BM or MPB as previously described,1 with minor modifications (see supplemental Methods for details). From weeks 4 to 9, ATOs were collected by adding magnetic-activated cell sorting buffer (phosphate-buffered saline with 0.5% bovine serum albumin and 2 mM EDTA) to each well and pipetting to dissociate the ATOs. Cells were then pelleted, resuspended in fluorescence-activated cell sorting buffer (phosphate-buffered saline with 2% fetal bovine serum), counted, and stained with the antibodies listed in supplemental Methods. Events were acquired on a BD LSR II Fortessa cell analyzer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software version 10.5.2 (Tree Star, Ashland, OR).
TCR-Vβ repertoire analysis and Gini-TCR skewing index calculation
The T-cell receptor-Vβ (TCR-Vβ) repertoire of mature T cells generated in vitro from a patient with DiGeorge syndrome (DGS) and from an ND was analyzed by flow cytometry using the IOTest β Mark TCR Repertoire Kit (IM3497, Beckman Coulter, Marseille, France). The cells were costained with anti-human CD45 V500, anti-human TCRαβ APC, and anti-human CD3 BV421 antibodies (see supplemental Methods for details) to identify the TCR-Vβ families in CD45+CD3+TCRαβ+ cells. Repertoires and their diversity were measured by using the Gini-TCR skewing index.2
Results
Figure 1A illustrates the strategy used to analyze in vitro T-cell maturation. As previously reported,1 one of the unique features of the ATO system is the ability to efficiently differentiate ND CD34+ cells into mature TCRαβ+CD3+ cells, thereby allowing detection of genetic defects that produce either early or late blocks in T-cell development.
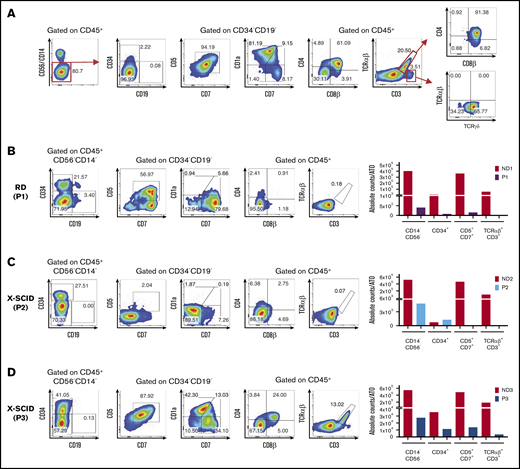
Human T-cell differentiation in ND samples and patients with early T-cell block. (A) Representative analysis of T-cell differentiation in an ND sample at 8 weeks (ND4). Cells were gated on LIVE/DEAD–CD45+CD14–CD56– cells to check for the presence of CD34+ and CD19+ cells and the expression of early and late T-cell commitment markers (CD5, CD7, CD1a, CD4, CD8α, CD8β, CD3, TCRαβ, and TCRγδ). (B-D) T-cell differentiation assay in patients with reticular dysgenesis (RD) (analyzed at 5 weeks) (B), and XSCID, carrying a null (P2) (6 weeks) (C), or a missense mutation (P3) (6 weeks) (D). The fluorescence-activated cell sorting (FACS) plots show expression of CD34, CD19, CD7, CD5, CD1a, CD4, CD8β, TCRαβ, and CD3 upon gating on LIVE/DEAD–CD45+CD14–CD56– cells. CD5, CD7, and CD1a plots are obtained upon gating on LIVE/DEAD–CD45+CD14–CD56–CD34– cells. The bar graphs show the absolute cell counts per ATO in the indicated gates in each patient’s sample and in the ND sample (red bars) analyzed in parallel in the T-cell differentiation assay.
Human T-cell differentiation in ND samples and patients with early T-cell block. (A) Representative analysis of T-cell differentiation in an ND sample at 8 weeks (ND4). Cells were gated on LIVE/DEAD–CD45+CD14–CD56– cells to check for the presence of CD34+ and CD19+ cells and the expression of early and late T-cell commitment markers (CD5, CD7, CD1a, CD4, CD8α, CD8β, CD3, TCRαβ, and TCRγδ). (B-D) T-cell differentiation assay in patients with reticular dysgenesis (RD) (analyzed at 5 weeks) (B), and XSCID, carrying a null (P2) (6 weeks) (C), or a missense mutation (P3) (6 weeks) (D). The fluorescence-activated cell sorting (FACS) plots show expression of CD34, CD19, CD7, CD5, CD1a, CD4, CD8β, TCRαβ, and CD3 upon gating on LIVE/DEAD–CD45+CD14–CD56– cells. CD5, CD7, and CD1a plots are obtained upon gating on LIVE/DEAD–CD45+CD14–CD56–CD34– cells. The bar graphs show the absolute cell counts per ATO in the indicated gates in each patient’s sample and in the ND sample (red bars) analyzed in parallel in the T-cell differentiation assay.
A summary of all patients analyzed and details on each ATO experiment performed can be found in supplemental Tables 1 and 2, respectively. Figure 1B illustrates representative fluorescence-activated cell sorting plots of T-cell development in patient 1 (P1) who had reticular dysgenesis resulting from mutations in the adenylate kinase 2 (AK2) gene.3,4 Consistent with previous observations that AK2 deficiency compromises survival of hematopoietic progenitor cells,5,6 the absolute count of CD45+CD14–CD56– cells recovered in culture was extremely low, and the few cells that survived were either blocked at CD34+ stage or were able to progress only up to Pre-T1 (CD5+CD7+) stage (Figure 1B).
We then tested CD34+ cells from P2 and P3 who carried distinct mutations in the IL2RG gene, which causes X-linked SCID.7 Figure 1C shows in vitro T-cell development in P2 who carried a null mutation. Decreased cell viability was detected starting with the earliest stages of differentiation, with most cells blocked at the CD34+ stage. Very few cells were able to progress to Pre-T1 stage (CD34–CD7+CD5+CD1a–). Conversely, generation of mature TCRαβ+CD3+ cells was observed in the sample from P3 who carried a hypomorphic IL2RG mutation associated with residual T-cell development in vivo (Figure 1D). Interestingly, the patient with reticular dysgenesis and the 2 patients with X-linked SCID retained a significant proportion of CD34+ cells even after 5 to 6 weeks of culture. As shown in supplemental Figure 1, upon gating on CD34+ cells from P1, P2, and P3 and from an ND analyzed at the same time of culture (6 weeks), we observed that P1 and P2 had a clear defect at early T precursor and Pro-T1 stages, whereas P3 had no clear defect in the differentiation of CD34+ cells (with a hypomorphic IL2RG mutation), consistent with generation of mature TCRαβ+CD3+ cells from CD34+ cells from this patient.
Next, we evaluated 6 patients with RAG mutations associated with SCID (P4 and P8), atypical SCID (P9), and combined immunodeficiency with granulomas (CID-G; P5-P7) (see supplemental Table 1). Results for 1 patient with SCID (P4) and 1 with CID-G (P6) are shown in Figure 2A-B. Surprisingly, in both patients, we observed that in all patients with RAG deficiency, T-cell development proceeded up to CD4+CD8+ double-positive (DP) stage. Moreover, longitudinal analysis of T-cell development demonstrated that the number of cells at the DP stage remained stable in P6 and P7 who had hypomorphic forms of RAG deficiency, whereas it declined over time in P8 and P9 who had SCID (supplemental Figure 2). In any case, none of the patients with RAG deficiency, irrespective of their clinical and immunologic phenotype, produced a sizeable number of mature TCRαβ+CD3+ cells (Figure 2A-B).
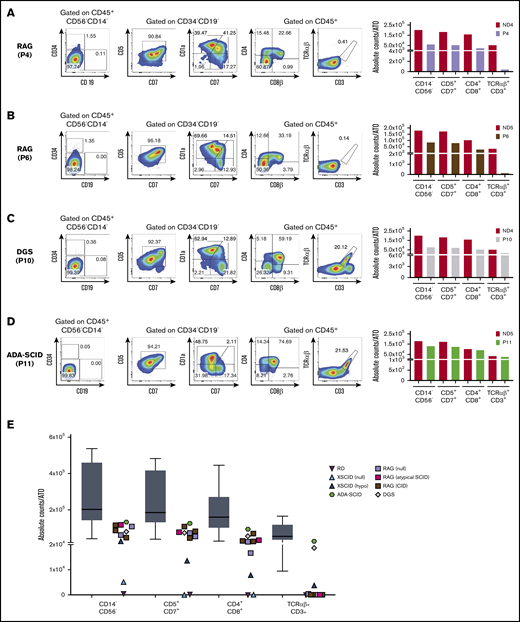
Human T-cell differentiation in patients with late T-cell block or without defects and summary of all T-cell differentiation assays. (A-D) T-cell differentiation assay in patients with RAG mutations, giving rise to a SCID (analyzed at 8 weeks) (A) or CID-G and autoimmunity (CID-G/AI) phenotype (6 weeks) (B), with DGS (8 weeks) (C), and with ADA-SCID (6 weeks) (D). The FACS plots show expression of CD34, CD19, CD7, CD5, CD1a, CD4, CD8β, TCRαβ, and CD3 upon gating on LIVE/DEAD–CD45+CD14–CD56– cells. CD5, CD7, and CD1a plots are obtained upon gating on LIVE/DEAD–CD45+CD14–CD56–CD34– cells. The bar graphs show the absolute cell counts per ATO in the indicated gates in each patient’ sample and in the ND sample (red bars) analyzed in parallel in the T-cell differentiation assay. (E) Summary of the absolute cell counts in the different T-cell subsets analyzed in all the experiments performed. The horizontal bar inside the gray box indicates the median, and the whiskers indicate 5% to 95% confidence intervals for all ND samples analyzed (n = 9). Each symbol represents a patient’s sample.
Human T-cell differentiation in patients with late T-cell block or without defects and summary of all T-cell differentiation assays. (A-D) T-cell differentiation assay in patients with RAG mutations, giving rise to a SCID (analyzed at 8 weeks) (A) or CID-G and autoimmunity (CID-G/AI) phenotype (6 weeks) (B), with DGS (8 weeks) (C), and with ADA-SCID (6 weeks) (D). The FACS plots show expression of CD34, CD19, CD7, CD5, CD1a, CD4, CD8β, TCRαβ, and CD3 upon gating on LIVE/DEAD–CD45+CD14–CD56– cells. CD5, CD7, and CD1a plots are obtained upon gating on LIVE/DEAD–CD45+CD14–CD56–CD34– cells. The bar graphs show the absolute cell counts per ATO in the indicated gates in each patient’ sample and in the ND sample (red bars) analyzed in parallel in the T-cell differentiation assay. (E) Summary of the absolute cell counts in the different T-cell subsets analyzed in all the experiments performed. The horizontal bar inside the gray box indicates the median, and the whiskers indicate 5% to 95% confidence intervals for all ND samples analyzed (n = 9). Each symbol represents a patient’s sample.
To investigate whether the ATO system can correctly identify the extra-hematopoietic nature of some forms of severe T-cell lymphopenia, we investigated T-cell development from CD34+ cells of a patient with a 22q.11.2 deletion, which caused complete DGS. As expected, we observed normal development of TCRαβ+CD3+ cells (Figure 2C). Moreover, the T cells generated in vitro expressed a broad range of TCR-Vβ families (supplemental Figure 3A), and the Gini-TCR skewing index was similar to that observed for TCRαβ+CD3+ cells generated in vitro from an ND and to what was observed for PB T cells from healthy controls (supplemental Figure 3B).
Finally, we observed normal T-cell differentiation in vitro in a patient with SCID as a result of adenosine deaminase (ADA) deficiency (Figure 2D). We speculate that normal in vitro T-cell development may reflect detoxification provided in trans by MS5-hDLL4 cells or it may reflect lower levels of toxic deoxyadenosine derivatives generated in the ATO system than in vivo in the thymus.8 In addition, because the ADA-SCID patient studied here was receiving enzyme replacement therapy when the BM sample was harvested, that treatment may have allowed in vivo detoxification of CD34+ cells and generation of mature T cells in vitro. The absolute count of cells at various stages of T-cell development per single ATO that were generated from patients and controls is shown in Figure 2E.
Because single ATOs may vary in their capacity to support T-cell development,1 it is important that multiple ATOs are collected and analyzed by flow cytometry at each time point. To demonstrate reproducibility of the assay, we compared the results obtained at various time points during culture of CD34+ cells from NDs and patients. As shown in supplemental Figure 4, consistent results were observed over time when multiple ATOs were collected and analyzed by flow cytometry at each time point.
Finally, the ATO system also allowed for the generation of a variable number of CD3–CD56+ natural killer (NK) cells (supplemental Figure 5). Virtually no CD56+ cells developed in vitro from CD34+ cells from the ADA-SCID patient, suggesting that the detoxification performed by the MS5-hDLL4 cells may not be sufficient to correct the defect in CD56+ cell generation. Similarly, no in vitro NK cell development was observed in the patient who carried the IL2RG null mutation, consistent with the critical role of γc-mediated signaling for NK cell development.9 By contrast, in vitro production of CD3–CD56+ NK cells was increased in patients with severe RAG deficiency (SCID, atypical SCID) compared with those with CID-G/AI, indicating a more pronounced unbalance between NK cell and T-cell development in the former.
Discussion
Our results demonstrate that the ATO system is a reliable tool for evaluating T-cell differentiation starting from BM/MPB CD34+ cells. The T-cell differentiation observed in vitro in the ATO system recapitulates well what was recently reported by single-cell RNA sequencing in human thymic samples.10 Moreover, the ATO system permits us to precisely define T-cell development blocks in patients with various forms of SCID and may reveal unexpected differences in data from mice. In particular, although Rag deficiency in mice causes a block at double-negative 3 stage,11,12 corresponding to the Pre-T-cell stage in humans, we observed that CD34+ cells from RAG-mutated patients were able to mature up to DP cells. These DP cells do not express CD3 on the cell surface (Figure 2A-B) and may therefore correspond to the DP CD3– cells described by Dik et al,13 and were recently renamed DP proliferating cells by Park et al.10 However, whether the DP cells generated in vitro from patients with RAG mutation express CD3 intracellularly remains to be defined. The observation that human RAG deficiency is associated with a late block in T-cell development may have important clinical implications. In particular, poor immune reconstitution has been reported after unconditioned hematopoietic stem cell transplantation for RAG deficiency.14,15 In the absence of conditioning, occupancy of thymic niches by RAG-mutant cells may cause competition between donor-derived and autologous cells up to the DP stage.
In this article, we describe our observation of a similar block in T-cell development at the DP stage in patients with RAG deficiency, irrespective of the severity of their clinical and immunologic phenotype. In a companion article, Bifsha et al16 reported a difference in the capacity of CD34+ cells from patients with RAG deficiency manifesting as SCID or Omenn syndrome to generate DP cells. We reported a similar loss of DP cells over time when co-culturing induced pluripotent stem cell–derived CD34+ cells from patients with RAG-null mutations with OP9-DLL4 cells, concurrent with an accumulation of single-strand DNA breaks and impaired cell survival.17 A similar mechanism may account for the lack of DP cells in patients with SCID and Omenn syndrome observed by Bifsha et al16 at week 5. In vivo, such decline in the number of DP cells could contribute to the lack of circulating TCRαβ+CD3+ cells in SCID patients. By contrast, survival of DP cells in CID-G patients with hypomorphic RAG mutations may be permissive for in vivo generation of TCRαβ+CD3+ cells, although less efficiently than normal.
Our observation that null IL2RG mutations are associated with a very early arrest in T-cell development are consistent with data obtained by Wiekmeijer et al18 in a xenograft transplantation model. Compared with that in vivo model, the ATO system offers the possibility of performing multiple analyses over time, starting from a very limited number of CD34+ cells, and permits us to observe the generation of mature T cells within 2 months of culture vs the 4 to 5 months necessary in animal studies. However, the ATO assay is limited to the study of the development of T and NK cells, so the xenograft transplantation model should be used to study human B-cell development.
By studying in vitro T-cell differentiation of CD34+ cells from a patient with complete DGS, we have demonstrated that the ATO system can be used to establish whether the severe T-cell lymphopenia observed in vivo is hematopoietic intrinsic or caused by a stromal (in this case, thymic) defect. This information is especially valuable for babies who are identified at birth as being positive to a newborn screening test for SCID, and in whom no definitive genetic causes of T-cell lymphopenia are identified. Determining whether their CD34+ cells have a normal capacity to differentiate to mature TCRαβ+CD3+ cells in the ATO system may guide treatment (eg, hematopoietic stem cell transplantation vs implantation of cultured thymus tissue).
Our results have identified some limitations of the ATO assay. In particular, we have observed that CD34+ cells from a patient with ADA deficiency were able to differentiate into TCRαβ+CD3+ cells. This finding may reflect detoxification provided in trans by the MS5-DLL4 cells. Alternatively, in vivo use of enzyme replacement therapy may have endowed CD34+ cells with the capacity to support T-cell differentiation in vitro. Whatever the mechanism, these results illustrate that the ATO assay cannot correctly identify metabolic causes of severe T-cell lymphopenia, and they indicate that appropriate enzymatic assays must be performed to identify these conditions.
Finally, although the use of BM/MPB CD34+ cells may represent a practical limitation of the assay, the system allows generation of a large number of T-cell progenitors that can be sorted and used for RNA sequencing studies to identify novel gene defects in patients with SCID of unknown genetic etiology.
Send data sharing requests to Luigi D. Notarangelo (luigi.notarangelo2@nih.gov).
Acknowledgments
This work was supported in part by the Intramural Research Program of the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant 1 ZIA AI001222-02), by the Bench to Bedside grant titled “RAG deficiency: From pathophysiology to precise gene editing” (L.D.N.), by the Italian Ministry of Health (PE-2-16-02363691) (A.V.), and by the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases and the Office of Rare Diseases Research, National Center for Advancing Translational Sciences, National Institutes of Health, cooperative agreement U54-AI082973 (L.D.N.).
Authorship
Contribution: M.B., F.P., A.V., and L.D.N. designed the experiments and analyzed the data; M.B., F.P., E.C., K.D., C.L.G., T.K., and N.S. performed the experiments; O.M.D., J.R.E.B., S.S.D., A.F.F., T.G., N.H., D.B.K., H.L.M., M.L.M., K.G.W., S.M.H., and O.M.D. provided patient samples; E.K.G. and D.D. helped gather informed consent forms and facilitated patient enrollment; C.S.S., A.M.-H., and G.M.C. contributed technical protocols and experimental guidance; M.B. and L.D.N. wrote the manuscript; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: M.L.M. is responsible for cultured thymus tissue implantation (CTTI; RVT-802), which is an investigational product implanted into patients under an Investigational New Drug application with the US Food and Drug Administration; is the sponsor of the investigations; and developed the technology for the CTTI. Duke University has licensed the technology to Enzyvant Therapeutics GmbH, and M.L.M. and Duke University have received royalties from Enzyvant. Enzyvant has funded portions of salaries for M.L.M. and her research team. G.M.C., C.S.S., and A.M.-H. are cofounders of PLUTO Immunotherapeutics, Inc, which has been granted an exclusive option to license the ATO technology from University of California, Los Angeles. The remaining authors declare no competing financial interests.
Correspondence: Luigi D. Notarangelo, Laboratory of Clinical Immunology and Microbiology, Immune Deficiency Genetics Section, Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health, 10 Center Dr, Building 10 CRC, Room 5W3950, MSC 1456, Bethesda, MD 20892-1456; e-mail: luigi.notarangelo2@nih.gov.