Key Points
BTK inhibitors revolutionized treatment; knock-in mice resistant to BTK inhibitors permit identification of new targets and side effects.
As proof of concept, we conclusively demonstrate that ibrutinib inhibits T cells irrespective of BTK.

Abstract
Pharmacological inhibitors of Bruton tyrosine kinase (BTK) have revolutionized treatment of B-lymphocyte malignancies and show great promise for dampening autoimmunity. The predominant BTK inhibitors tether irreversibly by covalently binding to cysteine 481 in the BTK catalytic domain. Substitution of cysteine 481 for serine (C481S) is the most common mechanism for acquired drug resistance. We generated a novel C481S knock-in mouse model and, using a battery of tests, no overt B-lymphocyte phenotype was found. B lymphocytes from C481S animals were resistant to irreversible, but sensitive to reversible, BTK inhibitors. In contrast, irreversible inhibitors equally impaired T-lymphocyte activation in mice, mimicking the effect of treatment in patients. This demonstrates that T-lymphocyte blockage is independent of BTK. We suggest that the C481S knock-in mouse can serve as a useful tool for the study of BTK-independent effects of irreversible inhibitors, allowing for the identification of novel therapeutic targets and pinpointing potential side effects.
Introduction
Bruton tyrosine kinase (BTK) inhibitors have greatly impacted treatment of B-cell malignancies by replacing unspecific chemotherapy regimens with targeted intervention.1 The first-generation oral BTK inhibitor ibrutinib (Imbruvica) has shown impressive clinical efficacy and is currently used as treatment of chronic lymphocytic leukemia, small lymphocytic lymphoma, mantle zone lymphoma, and Waldenström macroglobulinemia as well as for chronic graft-versus-host disease.2-4 Moreover, other B-cell tumors respond,5 and combining BTK inhibitors with compounds enhancing apoptosis seems particularly efficient.6 Ibrutinib binds covalently to the thiol group of cysteine (C) 481 in the adenosine triphosphate–binding site of BTK rendering the enzyme irreversibly inactive. This blocks B-cell receptor signal transduction, which is crucial for B-lymphocyte function, also in the absence of a foreign antigen.7,8 Similarly, the inhibitors acalabrutinib and zanubrutinib bind irreversibly to C481. All 3 have been approved by the US Food and Drug Administration (FDA), zanubrutinib as late as in November 2019.2,4,9-12
Genetic loss of functional BTK causes a primary immunodeficiency, X-linked agammaglobulinemia (XLA), which is clinically manifested as a selective B-lineage defect,13,14 even though BTK is also expressed in other hematopoietic lineages.15,16 Crucially, although ibrutinib, acalabrutinib, and zanubrutinib all bind and impair BTK’s activity, they also show both common and differential adverse effects, not seen in XLA patients. Among the reported side effects are diarrhea, headache, heart arrhythmias, increased blood pressure, thrombocyte malfunction with bleeds, and invasive fungal infections.17-19 The underlying mechanisms are still elusive even though binding of these compounds to other kinases has been identified.20,21
The therapeutic effect of ibrutinib during long-term follow-up is remarkable.22 Nevertheless, many patients with disease progression develop drug resistance.23,24 Unsurprisingly, C481 is the most commonly mutated BTK residue in cases of acquired resistance to ibrutinib.23-25 The predominating C481 mutation results in cysteine to serine (C481S) substitution, which abrogates covalent binding and profoundly reduces the efficacy of irreversible inhibitors.26,27 Critically, C481S BTK remains catalytically intact, and this replacement has been reported to even result in increased activity as compared with unmutated BTK.25,27,28
Apart from direct measurements of catalytic activity, there are other observations suggesting that the C481S substitution is compatible with full BTK activity.29 Thus, the C481S substitution has so far never been identified among XLA patients. In the international mutation repository, the BTKbase,30 with 1796 public variants including 917 unique forms (2019-09-04 version), none was caused by replacement of C481. Furthermore, insects naturally carry a serine residue in position 481 of their orthologous BTK, which is essential for fly development.31,32 We have previously genetically replaced Drosophila melanogaster Btk29A with human BTK and demonstrated that enzyme function is evolutionarily preserved.33-35
We here report the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas–mediated generation of mice carrying a C481S substitution in BTK. The edited enzyme was found to be fully active in biochemical assays, and, crucially, no overt phenotypic alterations were caused by this replacement. Furthermore, we demonstrate that the C481S substitution renders B-cell activation resistant to irreversible BTK inhibitors, whereas the off-target inhibition of T-lymphocyte activation remains unaffected. Collectively, this suggests that the gene-edited C481S mouse can serve as a tool to identify novel therapeutic targets as well as to discover off-target effects caused by irreversible BTK inhibitors in vivo.
Materials and methods
Animal studies
The C481S mutation was introduced into exon 15 of the mouse Btk gene (Ensembl gene ID: ENSMUSG00000031264 and NCBI gene ID: 12229) using CRISPR/Cas9-mediated gene editing (via zygote injection) with a specific single-guide RNA and an oligonucleotide (DNA template) carrying the modifications to be introduced. The targeting strategy was based on National Center for Biotechnology Information (NCBI) transcript NM_013482.2. The single-guide RNA was designed to be unique in GRCm38/mm10 (all potential off-target sequences had ≥3 mismatches). Mice were generated and maintained on a C57BL/6 background. Analyzed C481S mice and wild-type controls were sex and age matched. Most experiments were performed on 9- to 12-week-old mice. Autoantibody analysis was performed on 16-month-old animals whereas phenotypic fluorescence-activated cell sorter (FACS) analysis was carried out on both 9- to 12-week- and 20-month-old mice. All experiments were approved by the local ethics committee.
Flow cytometry
Dissected organs (bones, spleen, and thymus) were crushed in phosphate-buffered saline (PBS) with 2% fetal calf serum (FCS) and filtered through a 70-μm nylon filter. For preparation of peripheral blood (PB) cells, red blood cells were removed using 1% dextran, remaining RBCs were lysed using 0.8% NH4Cl, and remaining cells resuspended in PBS with 2% FCS. Prepared cells were subsequently Fc-blocked using anti-CD16/32 (93; BD Biosciences) and stained with fluorochrome-conjugated antibodies (Abs) as previously described.36 For Ab panels used, see supplemental Table 1. Propidium iodide was used to discriminate dead cells. Data were acquired on a FACSAria IIu (BD Biosciences) and data analysis using Flowjo 9.9.6.
Immunohistochemistry
Spleens were frozen in cryostat-embedding medium (Bio-Optica), and 8-mm-thick sections were cut using a cryostat microtome. After overnight drying, the slides were fixed in acetone. Before staining, slides were blocked with 5% goat serum (Dako Cytomation) in PBS, and then stained with the fluorophore-conjugated Abs and biotin Ab at 4°C overnight. Afterward, the sections were washed and subsequently incubated with fluorophore-conjugated streptavidin for 1 hour at room temperature. The reagents used for immunohistochemistry were biotin-conjugated anti-B220 (RA3-6B2; BioLegend), fluorescein isothiocyanate–conjugated anti-mouse CD169 (MOMA-1, Bio-Rad Abs), Alexa Fluor 647 streptavidin (BioLegend), and allophycocyanin-conjugated anti–T-cell receptor β (TCRβ) (clone H57-597; BioLegend). Slides were mounted with ProLong Diamond Antifade Mountant (Thermo Fisher Scientific). Visual data were acquired with a confocal microscope (Zeiss LSM880) and recorded with LSM Image software.
ELISA
For total immunoglobulin G (IgG), IgM, and IgG3 enzyme-linked immunosorbent assay (ELISA), serum was diluted at 1/1000 to 1/128 000, and incubated for 1 hour at 37°C on plates coated overnight at 4°C with 2.5 μg/mL anti-IgG (H+L) in PBS. Then, plates were incubated for 1 hour at 37°C with horseradish peroxidase (HRP)–conjugated goat Ab to mouse IgG, IgM, and IgG3, developed with 3,3′,5,5′-tetramethylbenzidine (TMB) substrate sets, and blocked with sulfuric acid (1 M); results were measured at 450 nm with a Bio-Rad microplate reader. The reagents used for the ELISA were anti-IgG (H+L) (Biosite), TMB Super Sensitive (BioLegend), the polyclonal Abs goat anti-mouse IgG-HRP, goat anti-mouse IgM-HRP, and goat anti-mouse IgG3-HRP, purchased by Southern Biotech. For anti-DNA reaction, 50 μL per well of serum was incubated for 1 hour at 37°C on plates coated overnight at 4°C with 5 μg/mL methylated bovine serum albumin in PBS and 50 μg/mL calf thymus DNA in PBS (Sigma-Aldrich). Then, plates were incubated for 1 hour at 37°C with anti-mouse immunoglobulin-HRP and treated as mentioned previously.
BTK inhibitors
BTK inhibitors used were purchased from: Selleckchem (ibrutinib and acalabrutinib), Chemgood (zanubrutinib), and MedChemTronica (RN486). BTK inhibitors were dissolved in dimethyl sulfoxide and diluted in the medium prior to each experiment. For in vivo ibrutinib treatment, mice were (for 1 week) given drinking water with ∼0.3 mg/mL ibrutinib and 3% (2-Hydroxypropyl)-β-cyclodextrin (H107; Sigma-Aldrich).
Western blot analysis
Purified B lymphocytes were obtained from spleen by using negative selection (EasySep mouse B-cell enrichment kit; StemCell Technologies). After isolation, B lymphocytes were starved in serum-free Iscove modified Dulbeco medium (IMDM) with 50 μM β-mercaptoethanol (Gibco/Life Technologies) for 1 hour. During starvation, cells were treated with ibrutinib (0.5 μM) or RN486 (1 μM). Subsequently, the cells were activated for 5 minutes at room temperature, with H2O2 (4 mM) and anti-mouse IgM (10 μg/mL, 1022-01; Southern Biotech). Whole-cell lysates and immunoblotting analysis were performed as described previously.28 The following Abs were used: anti-actin (A5441; Sigma-Aldrich), anti-BTK (270-284; Sigma-Aldrich), anti-BTK pY551 (24a/BTK; BD Biosciences), anti-BTK pY223 (EP420Y; Abcam), anti–phospholipase C γ2 (PLCG2) (rabbit polyclonal; Biotech), and anti-PLCG2 pY753 (polyclonal; Abcam). The secondary Abs, goat anti-mouse 800CW, goat anti-rabbit 800CW, goat anti-mouse 680LT, and goat anti-rabbit 680 were purchased from LI-COR Biosciences GmbH. Membrane scanning was performed in the Odyssey Imager from LI-COR Biosciences GmbH. The signals of total and phosphorylated proteins were quantified by the densitometric program NIH ImageJ 1.52a.
For western blot analysis of cells from ibrutinib-treated animal, fresh splenocytes were obtained and starved in serum-free IMDM with 50 μM β-mercaptoethanol for 2 hours. Subsequently, cell activation and immunoblotting were carried out as described in the paragraph above.
Cell stimulation, inhibition, and cell-viability assay
For B-lymphocyte activation, 2 × 106 splenocytes were seeded in 0.4 mL of IMDM containing 15% FCS and 50 μM β-mercaptoethanol per well in a 24-well plate. Anti-mouse IgM 20 μg/mL (1022-01; Southern Biotech) was added to each well. For T-lymphocyte activation, the 24-well plates were coated with anti-CD3e (145-2C11; eBioscience) according to the manufacturer’s protocol. Subsequently, 2 × 106 splenocytes were added to each well in 0.4 mL of RPMI 1640 medium (Sigma-Aldrich) with 10% FCS. Five minutes after B- or T-lymphocyte activation, BTK inhibitor was added and the cells were cultured for 30 hours at 37°C. To analyze stimulation responses, cells were harvested and stained with fluorochrome-conjugated Abs (supplemental Table 1) in Annexin V staining buffer (BioLegend). LIVE/DEAD Aqua (Invitrogen) was used for dead cell discrimination. Analysis was performed on FACSAria IIu (BD Biosciences) and data analysis using Flowjo 9.9.6 software (Flowjo).
Results
To investigate the potential effect of BTK with cysteine 481 to serine substitution (C481S) on hematopoietic development and to generate a model to identify new drug targets, we used CRISPR-Cas9 to introduce the C481S substitution into the mouse Btk gene by exchanging a TGC for an AGT codon (supplemental Figure 1). In addition to generating the sequence-verified C481S substitution, silent mutations were introduced to create an MscI restriction site for analytical purposes, and to prevent further Cas9 activity on the already targeted locus (supplemental Figure 1).
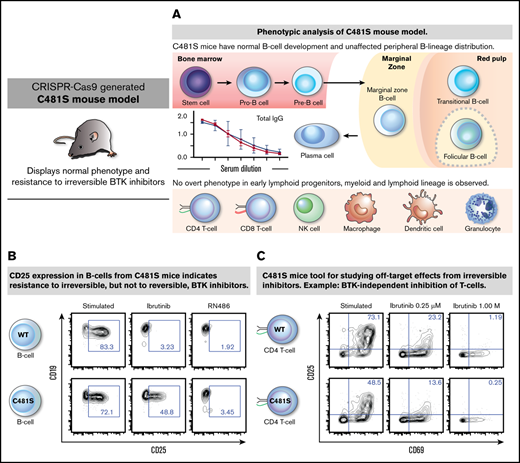
Mice carrying C481S in the Btk gene show normal B-cell development and generation of peripheral B-cell subsets
We first set out to determine whether the C481S substitution impacted on the B-cell lineage. In order to identify aberrations in B-cell development and the generation of peripheral B-cell subsets, we analyzed B-lymphocyte progenitors in the bone marrow (BM) as well as peripheral B cells in spleen and peritoneal cavity (PeC) by flow cytometry (Figure 1 left panels). We did not detect any significant differences in the analyzed B-cell population in 9- to 12-week-old mice in terms of total cellularity (in BM or spleen) or distribution of B-cell subsets (PeC) (Figure 1 right panels). This clearly suggests that both B-cell development and peripheral maturation of B lineage cells are unaffected by the C481S substitution.
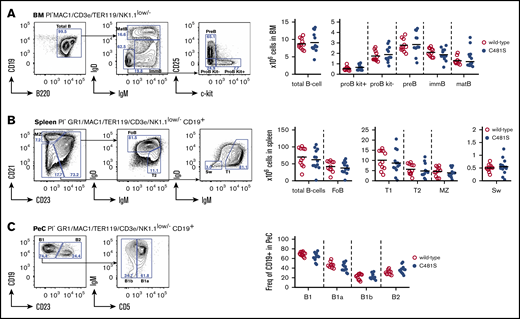
Bone marrow B-cell development and generation of peripheral B-cell subsets is normal in C481S-mutant animals. (A-C, left) Gating strategy for identification of B-cell developmental stages and peripheral subsets in indicated organs. Prior gating is indicated above the FACS plots. Propidium iodide (PI) was used for dead cell discrimination. (A-B, right) Absolute numbers of total B cells and B-cell subsets in bone marrow (BM) and spleen. (C, right) Frequency of subsets within B cells in the peritoneal cavity (PeC). (A-C, right) Each dot represents data from individual animals; vertical bars indicate mean and 2-tailed Mann-Whitney U test was used to calculate significance. FoB, follicular B cell; immB, immature B cell; matB, mature B cell; MZ, marginal zone B cell; Sw, switched B cell; T1, transitional 1 B cell; T2, transitional 2 B cell.
Bone marrow B-cell development and generation of peripheral B-cell subsets is normal in C481S-mutant animals. (A-C, left) Gating strategy for identification of B-cell developmental stages and peripheral subsets in indicated organs. Prior gating is indicated above the FACS plots. Propidium iodide (PI) was used for dead cell discrimination. (A-B, right) Absolute numbers of total B cells and B-cell subsets in bone marrow (BM) and spleen. (C, right) Frequency of subsets within B cells in the peritoneal cavity (PeC). (A-C, right) Each dot represents data from individual animals; vertical bars indicate mean and 2-tailed Mann-Whitney U test was used to calculate significance. FoB, follicular B cell; immB, immature B cell; matB, mature B cell; MZ, marginal zone B cell; Sw, switched B cell; T1, transitional 1 B cell; T2, transitional 2 B cell.
Mice carrying C481S in the Btk gene show normal B-cell follicle organization
In order to further confirm that the substitution is not affecting the B-cell spatial distribution or the structure of peripheral lymphoid organs, we performed immunohistochemical analysis of the spleen and lymph nodes. We found that the splenic follicular structure was maintained with well-defined B- and T-lymphocyte areas and metallophilic macrophages showed normal distribution along the marginal sinus lining the follicles (Figure 2A). Furthermore, the relative structure of the spleen and the size of white vs red pulp areas was intact. In the lymph nodes from C481S mice we found a normal spatial distribution with central T-cell areas and round B-cell follicles near the subcapsular part of the node (Figure 2B).
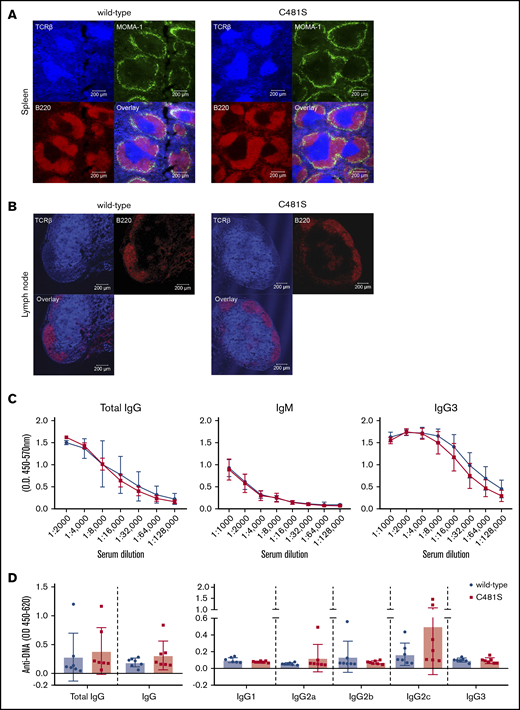
The structure of peripheral lymphoid organs and serum immunoglobulin levels are not affected by the C481S substitution. (A-B) Immunofluorescence staining of spleen and lymph nodes sections. Labeled Abs against B220 (red), MOMA-1 (green), and TCRβ (cyan) were used for spleen and B220 (red) and TCRβ (cyan) for lymph nodes. Scale bar, 200 μm. (C) ELISA was performed for total IgG, IgM, and IgG3; serum from 5 wild-type and 5 C481S mice was diluted to indicated concentrations. (D) Anti-DNA total IgG, IgM, and IgG subtypes Abs were measured by ELISA. Each dot represents data from individual animals, error bars indicate standard deviation (SD) of the mean and unpaired 2-tailed Mann-Whitney U test was used to calculate significance.
The structure of peripheral lymphoid organs and serum immunoglobulin levels are not affected by the C481S substitution. (A-B) Immunofluorescence staining of spleen and lymph nodes sections. Labeled Abs against B220 (red), MOMA-1 (green), and TCRβ (cyan) were used for spleen and B220 (red) and TCRβ (cyan) for lymph nodes. Scale bar, 200 μm. (C) ELISA was performed for total IgG, IgM, and IgG3; serum from 5 wild-type and 5 C481S mice was diluted to indicated concentrations. (D) Anti-DNA total IgG, IgM, and IgG subtypes Abs were measured by ELISA. Each dot represents data from individual animals, error bars indicate standard deviation (SD) of the mean and unpaired 2-tailed Mann-Whitney U test was used to calculate significance.
In line with the unaltered naive B-cell populations and intact structure of secondary lymphoid organs we found comparable levels of serum Abs in C481S and wild-type mice, with no significant differences in total IgG and IgM levels (Figure 2C). A hallmark of BTK deficiency in mice is severely reduced IgG3 production.37 However, our results show that C481S mice do not have reduced IgG3 because IgG3 levels are similar to those found in wild-type mice, which is also in line with unaltered numbers of marginal zone (MZ) B cells that have been shown to be important for production of this subclass (Figure 2C).38
C481S mice display no alterations in hematopoietic progenitors or in the generation of non–B-lineage leukocytes
With the B- and T-cell zones in spleen appearing normal, we next verified that early lymphoid progenitors and T-cell development were unaffected by the C481S substitution in 9- to 12-week-old mice. We did not identify any significant changes in hematopoietic stem cells (HSCs), lymphoid-primed multipotent progenitors (LMPPs), or in common lymphoid progenitor (CLP) subsets that precede the proB-cell stage in BM39-41 (supplemental Figure 2A). Nor did we observe any differences in thymic T-cell development42 (supplemental Figure 2B). In line with T-cell development being unaffected, no differences were found in the C481S mice in terms of CD4+ and CD8+ T cells in spleen and PB (supplemental Figure 3A-B). Further, as BTK is expressed in other mature leukocytes, including macrophages, we analyzed macrophages, granulocytes, dendritic cells and natural killer cells in spleen and PB but, similarly, we found no significant changes in the C481S animals (supplemental Figure 3A-B). Taken together, this shows that the C481S substitution has no detectable effect on the B-cell developmental pathway and that hematopoiesis is overall normal.
No major phenotype changes were observed in old C481S animals
The natural Ab repertoire is in part polyreactive with affinity for DNA.43 Hence, a dysregulation of B-cell activation could potentially lead to natural Abs inducing autoimmune disease in aged mice due to deficiency in B-cell tolerance checkpoints. To investigate this, we measured anti-DNA reactivity in mice that were ∼16-month-old but did not find significant differences in C481S mice compared with controls (Figure 2D).
Furthermore, in line with the results obtained from young animals (9-12 week-old), 20-month-old mice did not show differences in the peripheral B-cell subsets from spleen and PeC, when compared with age matched wild-type mice (supplemental Figure 4A-B). Plasmocytoid dendritic cell numbers in spleen increased more in aged C481S mice as compared with the normal controls. The frequency of B cells in peripheral blood remained the same in aged C481S animals but were slightly increased among healthy controls (supplemental Figure 4A-D). Collectively, this suggests that there are no major differences in hematopoietic cell development in C481S knock-in mice.
Histopathological analysis was also carried out for heart, kidney, liver, and lung in 20-month-old mice (supplemental Table 2). Although, as expected, age-related changes were observed, there were no significant differences between wild-type and C481S mice.
Reversible, but not irreversible, inhibitors affect BTK-signaling–induced activation and viability of C481S B lymphocytes
Previous reports have shown that reversible inhibitors potently inhibit BTK C481S in transfected cells.44,45 To confirm this finding ex vivo in the endogenous protein, we evaluated BTK´s catalytic activity by measuring the phosphorylation of tyrosine (Y) 223 and Y551 in BTK as well as Y753 in the downstream substrate PLCG27,8,46 (Figure 3). The irreversible BTK inhibitor ibrutinib, as expected,28 blocked phosphorylation of BTK Y223 and PLCG2 Y753 but not that of BTK Y551 in wild-type B lymphocytes. Contrary to this, ibrutinib-treated B lymphocytes isolated from the C481S mice displayed minimal effect on BTK and PLCG2 phosphorylation (Figure 3), hence, confirming that C481S renders C481 targeting by irreversible inhibitors ineffective. However, as the reversible inhibitors interact with BTK in a different manner,45 their activity consequently may not be affected by the C481S substitution. In agreement with this, we found that the reversible inhibitor RN486 binding to the BTK kinase domain through hydrogen bonds to the lysine 430, threonine 474, and glycine 414 residues,27 has similar inhibitory capacity in B cells obtained from both wild-type and C481S mice (Figure 3A). To confirm this in vivo, mice were given (2-Hydroxypropyl)-β-cyclodextrin-solubilized ibrutinib (0.3 mg/mL) in the drinking water for 1 week. As expected, this confirmed that the irreversible BTK inhibitor only blocks phosphorylation of BTK Y223 and PLCG2 Y753 in wild-type animals (Figure 3B). One C481S mouse deviated from this pattern and displayed some sensitivity to ibrutinib. We do not know the reason for this, but, potentially, excessive consumption of drinking water causing increased uptake of ibrutinib may have contributed.
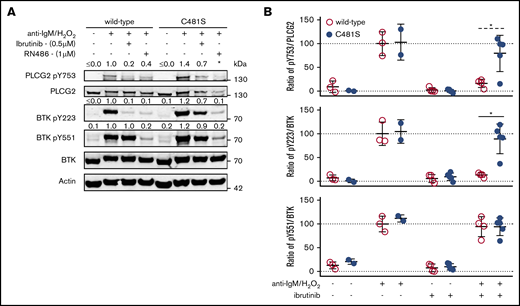
BTK phosphorylation is minimally affected by ibrutinib treatment in B cells from C481S mice. (A) Western blot on whole-cell lysates from B cells with indicated genotype (wild-type or C481S), inhibitor treatment (ibrutinib or RN486) and stimulation (anti-IgM and H2O2). Numbers above bands indicate densiometric quantification. *Value could not be calculated. (B) Ratio of phosphorylated protein over total protein (as quantified by densiometric analysis) from splenocytes with indicated genotype, in vivo ibrutinib treatment, and in vitro stimulation (anti-IgM and H2O2). For in vivo ibrutinib treatment, mice were given ibrutinib (0.3 mg/mL) in the drinking water for 1 week. Each dot represents data from an individual animal. Error bars indicate mean and SD; the unpaired 2-tailed Mann-Whitney U test was used to calculate significance (*P < .05); (dashed line) indicates significance calculated after exclusion of the outlier. For densiometric quantification in panels A-B, background signal was subtracted, β-actin used as an internal loading control and values displayed normalized to the mean protein ratio in wild-type cells.
BTK phosphorylation is minimally affected by ibrutinib treatment in B cells from C481S mice. (A) Western blot on whole-cell lysates from B cells with indicated genotype (wild-type or C481S), inhibitor treatment (ibrutinib or RN486) and stimulation (anti-IgM and H2O2). Numbers above bands indicate densiometric quantification. *Value could not be calculated. (B) Ratio of phosphorylated protein over total protein (as quantified by densiometric analysis) from splenocytes with indicated genotype, in vivo ibrutinib treatment, and in vitro stimulation (anti-IgM and H2O2). For in vivo ibrutinib treatment, mice were given ibrutinib (0.3 mg/mL) in the drinking water for 1 week. Each dot represents data from an individual animal. Error bars indicate mean and SD; the unpaired 2-tailed Mann-Whitney U test was used to calculate significance (*P < .05); (dashed line) indicates significance calculated after exclusion of the outlier. For densiometric quantification in panels A-B, background signal was subtracted, β-actin used as an internal loading control and values displayed normalized to the mean protein ratio in wild-type cells.
Because, during the in vitro experiments, ibrutinib is constantly present (Figure 3A), we also used a washout procedure to better reflect in vivo conditions (supplemental Figure 5). In these experiments, the effect of an ibrutinib concentration of 1 or 4 μM was studied. Although cells from C481S mice were affected by 4 μM, a concentration never reached in treated patients, following washout they were no longer inhibited. In contrast, cells from wild-type mice were sensitive to 1 μM ibrutinib during washout conditions. This again confirms that the C481S alteration confers resistance to irreversible BTK inhibitors.
BTK is important for the survival of peripheral B cells and apoptosis is induced in the absence of functional BTK.13,14,47,48 Similarly, apoptosis is induced in B cells upon treatment with either irreversible or reversible BTK inhibitors.1,27,44 In order to investigate if BTK-inhibitor–induced cell death is reduced as a consequence of the C481S substitution, we characterized the viability of B cells following stimulation in the presence or absence of the inhibitors. To this end, B cells from wild-type, C481S and BTK knockout (KO) mice were stimulated in vitro with anti-IgM and treated with irreversible (ibrutinib, acalabrutinib and zanubrutinib) or reversible (RN486) BTK inhibitors. Two concentrations of irreversible BTK inhibitors were used, corresponding to half and double the physiological peak concentration measured in the serum of the treated patients.8,11,49 As expected,50 cell death was observed in both unstimulated and stimulated B cells from BTK KO mice (with or without treatment with inhibitors) whereas wild-type cells survived well in the absence of inhibitors (Figure 4A). In contrast, we observed that B cells from C481S mice survived treatment with irreversible BTK inhibitors (ibrutinib and zanubrutinib) even at high concentrations whereas acalabrutinib induced cell death when the highest tested concentration (3 μM) was used (Figure 4A). Moreover (Figure 3), we found that the reversible inhibitor RN486 considerably reduced the viability of B cells carrying the C481S substitution (Figure 4A).
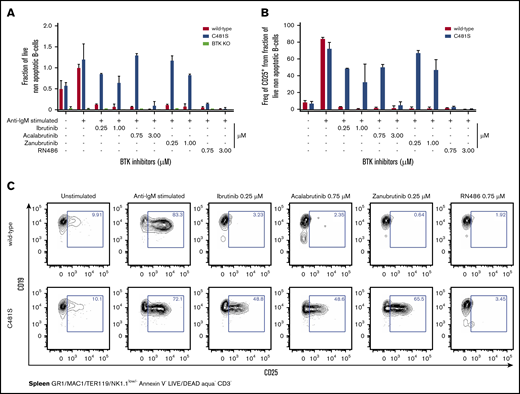
Reversible, but none of the irreversible, BTK inhibitors affect viability or blocks CD25 expression in isolated B cells from C481S. (A) Viability of B cells as determined by flow cytometry following indicated stimulation and BTK-inhibitor treatment. Cell viability was assessed using annexin V and LIVE/DEAD (viability marker) staining. (B) CD25 expression on B cells as determined by flow cytometry following indicated stimulation and BTK inhibitor treatment. (C) Representative data from 1 of 2 independent experiments. Prior gating is indicated below FACS plots. (A-B) Bars indicate mean with SD. Data are from 2 independent experiments and a total 4 wild-type, 4 C481S, and 2 BTKKO mice were analyzed. (A) Values were normalized to the percentage of live (LIVE/DEAD− Annexin V−) cells in the average of wild-type stimulated controls from each independent experiment. Data from BTK KO mice are only shown in panel A. The highest dose of acalabrutinib reduced survival and CD25 expression in C481S mice.
Reversible, but none of the irreversible, BTK inhibitors affect viability or blocks CD25 expression in isolated B cells from C481S. (A) Viability of B cells as determined by flow cytometry following indicated stimulation and BTK-inhibitor treatment. Cell viability was assessed using annexin V and LIVE/DEAD (viability marker) staining. (B) CD25 expression on B cells as determined by flow cytometry following indicated stimulation and BTK inhibitor treatment. (C) Representative data from 1 of 2 independent experiments. Prior gating is indicated below FACS plots. (A-B) Bars indicate mean with SD. Data are from 2 independent experiments and a total 4 wild-type, 4 C481S, and 2 BTKKO mice were analyzed. (A) Values were normalized to the percentage of live (LIVE/DEAD− Annexin V−) cells in the average of wild-type stimulated controls from each independent experiment. Data from BTK KO mice are only shown in panel A. The highest dose of acalabrutinib reduced survival and CD25 expression in C481S mice.
Next, we investigated the effects of the C481S substitution on B-cell activation using surface CD25 (interleukin-2 receptor α-chain) expression as a marker of activation.51 In agreement with survival and BTK phosphorylation data for C481S B cells, we found no major effect of the irreversible inhibitors, whereas RN486 completely blocked CD25 expression. In contrast, wild-type cells failed to display CD25 after stimulation regardless of which inhibitor that was used (Figure 4B-C). Expression of another activation marker, CD69 was also analyzed and the results were similar as for CD25 (supplemental Figure 6A-B).
Taken together, we conclude that BTK C481S confers resistance to irreversible BTK inhibitors and maintains the activity of the BTK protein needed for proper B-cell function.
The C481S mouse in vivo model for studying BTK-independent effects caused by irreversible BTK inhibitors
As shown, the C481S mouse model is resistant to irreversible inhibitors at relevant physiological concentrations. Therefore, any observed treatment effect is expected to result from binding of the inhibitors to other proteins. In C481S mice, tethering to other kinases with a cysteine in the active site would likely be the most common consequence of treatment with irreversible BTK inhibitors. In addition to BTK, 9 kinases have been previously shown to have a cysteine residue in the adenosine triphosphate–binding site.20 These include the IL-2-inducible kinase (ITK), which has a cysteine at the position 442 and is a key transducer for T-cell receptor-mediated activation and proliferation of T cells.52 We investigated activation of CD4+ and CD8+ T cells in C481S mice and wild-type controls using CD69 and CD25 as activation markers. We observed a reduction of >50% in CD25/CD69 coexpression in stimulated CD4+ and CD8+ T lymphocytes treated with 0.25 μM ibrutinib regardless of mouse genotype (Figure 5). Moreover, we observed a total lack of CD25 expression in activated CD4+ and CD8+ T cells after treatment with 1.0 μM ibrutinib in both wild-type and C481S mice (Figure 5). Hence, ibrutinib displays significant BTK-independent effects on the T-cell lineage. This is in agreement with previous studies reporting that T-cell activation is blocked by irreversible binding of ibrutinib to ITK,53 although in these investigations an effect on BTK expressed by T cells could not be completely ruled out. Collectively, this demonstrates that the C481S knock-in mouse can provide a powerful system for the study of irreversible kinase inhibitors and identification of new therapeutic targets.
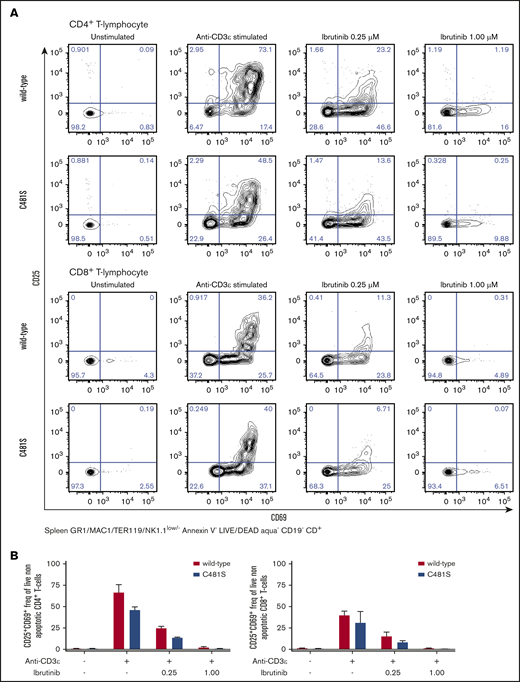
Ibrutinib blocks CD4+ and CD8+ T-cell activation in anti-CD3 stimulated T cells in a BTK-independent manner. (A-B) T lymphocytes obtained from spleen of indicated genotypes were stimulated with anti-CD3ε Abs in the presence of indicated concentrations of ibrutinib and subsequently analyzed by flow cytometry for the activation markers CD69 and CD25. Representative data from 1 of 2 independent experiments. (A) Prior gating is indicated below the FACS plots. (B) Bars indicate mean with SD from 2 biological replicates.
Ibrutinib blocks CD4+ and CD8+ T-cell activation in anti-CD3 stimulated T cells in a BTK-independent manner. (A-B) T lymphocytes obtained from spleen of indicated genotypes were stimulated with anti-CD3ε Abs in the presence of indicated concentrations of ibrutinib and subsequently analyzed by flow cytometry for the activation markers CD69 and CD25. Representative data from 1 of 2 independent experiments. (A) Prior gating is indicated below the FACS plots. (B) Bars indicate mean with SD from 2 biological replicates.
Discussion
Using CRISPR-Cas–mediated gene editing we have generated a mouse carrying a cysteine to serine substitution at residue 481 (C481S) of BTK. With the disulfide-forming cysteine 481 residue in the BTK catalytic SH1 domain replaced with a serine, this has been shown to render the modified BTK resistant to several irreversible BTK inhibitors. Hypothetically, irreversible BTK inhibitors could still bind in a noncovalent manner and therefore binding efficiency of new compounds to C481S needs to be determined. The engineered BTK C481S mouse was found to carry an enzymatically active form of BTK, and we observed no overt phenotypic changes. Among patients who develop resistance to irreversible inhibitors, the C481S replacement predominates, being an order of magnitude more common as compared with other substitutions, such as C481G (glycine), F (phenylalanine) R (arginine), Y (tyrosine), and W (tryptophan), which are catalytically less active,23-29 and hence do not activate downstream signaling pathways.54,55 Therefore, the high frequency of C481S mutations is not surprising, considering that this substitution allows B cells to maintain the critical BTK signaling while at the same time providing resistance to therapy. Neither is it unexpected that this mutation would be present prior to inhibitor treatment,56 as it has no negative influence on the carrier B cell.
Loss-of-function BTK mutations in mice cause an X-linked immunodeficiency (Xid) phenotype, which in the BM is characterized by a modest arrest at the proB stage of B-cell development, with an increase of the proB-cell population and concomitant decrease in the number of immature and mature B cells.47,50 In the spleen of Xid mice there is a 30 to 40% reduction of IgM+ B cells and mature IgMlo/IgDhi B cells are reduced almost by half.47,50,57 There is a lack of CD5+ B1a cells and reduction of total IgM+ B1 cells in the peritoneal cavity of Xid mice.47,50 In aged, 20-month-old C481S mice there were some slight changes in the frequency of B cells and plasmocytoid dendritic cells as compared with younger mice. The significance of this, if any, remains unknown and needs to be investigated in larger cohorts. Instead of a reduction of all immunoglobulin classes as seen in XLA patients,58,59 mice lacking the endogenous BTK enzyme have reduced IgM and very low levels of IgG3.47,50,57 In contrast, the C481S mouse has normal immunoglobulins, including IgG3. Autoimmune manifestations are a frequent finding in aged mice with dysregulated immunoglobulin production.60,61 Again, in line with normal signaling function of BTK C481S, we did not observe any indication of such abnormalities; instead the level of anti-DNA Abs was similar in wild-type and knock-in mice.
The first clinically approved BTK inhibitor, ibrutinib, received its first FDA-approval in 2013. Since then many patients have been treated worldwide and there is now vast experience from adverse effects not caused by impairment of B cells.49,53,62 Thus, such phenomena are not observed in XLA patients, but are instead presumed to be due to non–BTK-related adverse effects caused by ibrutinib or other BTK inhibitors. To this end, a population of cells, whose clinical importance has been increasingly appreciated, is the one forming the tumor microenvironment, which seems essential for leukemia and lymphoma development.63 Whereas these cells in B-cell malignancies by definition are non-B cells, many of them express BTK and could therefore represent a potentially important target for BTK inhibitors. We have initiated further studies in which the C481S mutation will be transferred to transgenic mice prone to develop chronic lymphocytic leukemia. This will allow us to study the interaction between malignant B cells resistant to irreversible BTK inhibitors and the tumor microenvironment.
Similarly, it has also been reported that leukemia patients under ibrutinib treatment show impairment of T-cell function.53 For this reason, we studied the effect of ibrutinib on T cells derived from the C481S mouse. Upon activation C481S T cells were in fact sensitive to ibrutinib, conclusively demonstrating that the impairment is BTK-independent and instead likely reflects an off-target inhibition of ITK. In a similar way, the mechanisms underlying off-target effects like atrial fibrillation or bleeding could be studied in a BTK-independent manner using the C481S mouse model.
For many medicines new indications are found because the drug causes unanticipated effects. An example of this is the first approved tyrosine kinase inhibitor, imatinib, which was developed to target the catalytic region in the BCR-ABL fusion protein.64 It was soon realized that mesenchymal gastrointestinal stromal tumors (GISTs), caused by activating mutations of the KIT gene,65 also respond to this compound.66 Today imatinib, which binds to the KIT receptor tyrosine kinase, has become the standard, first-line treatment of advanced/metastatic GIST. In a similar way it is possible that irreversible BTK inhibitors could have effects on non–B-lineage tumors by tethering to other kinases than BTK. Moreover, binding may not be restricted to kinases and the effect may not be limited to tumors. In all of these cases, the C481 mouse represents a useful model for deciphering the underlying events.
Researchers requesting original data and the gene-edited knock-in mouse strain should contact the corresponding author, C. I. Edvard Smith, at edvard.smith@ki.se.
Acknowledgments
The authors are indebted to Aleksandra Krstic for managing the FACS facility at the Center for Cell Analysis hosted at the Karolinska University Hospital. The authors thank Birger Christensson, Karolinska University Hospital, for expert assistance in the histopathological analysis of tissues.
This work was supported by grants from the Swedish Cancer Society and the Swedish County Council (ALF Project) (C.I.E.S.); a grant from the Swedish Medical Research Council (C.I.E.S. and R.Z.); and grants from the Swedish Cancer Society, the Swedish Research Council, the Foundation for Strategic Research, the Knut and Alice Wallenberg Foundation, and a generous donation from Björn and Lena Ulvaeus (R.M.). H.Y.E. acknowledges COLCIENCIAS scholarship program 756 (2016). M.C.I.K acknowledges grants from the Swedish Cancer Society and Swedish Research Council. O.S. was supported by a grant from the Egyptian Ministry of Higher Education. L.Y. acknowledges grants from the Science and Technology Fund of Jiangsu Commission of Health (grant no. H2018085), the “333 Projects” Foundation of Jiangsu Province (grant no. BRA2017243), and the “533 Projects” Foundation of Huai’an City (grant no. HAA201739).
Authorship
Contribution: C.I.E.S. perceived the project and proposed the generation of the mouse model; R.M. and C.I.E.S. conceptualized the project, designed the work, and obtained the mouse model; H.Y.E., T.B., and R.M. performed phenotyping by flow cytometry, viability test, and B- and T-cell stimulations; H.Y.E. performed western blot for kinase activity and in vivo experiments; A.B. prepared and coordinated animal experiments; C.H. and M.C.I.K. carried out immunofluorescence stainings, ELISA, and autoantibody assays; D.G. prepared mouse samples; O.S. prepared solutions with solubilized ibrutinib in the drinking water; M.Á.D.C. carried out the histopathological analysis; H.Y.E., T.B., A.B., C.H., L.P.-P., M.C.I.K., and C.I.E.S. analyzed and interpreted results; L.P.-P. performed statistical analyses; R.Z. interpreted the results and assisted in obtaining the mouse model; L.Y. assisted in obtaining the mouse model; T.B., C.H., and M.C.I.K. wrote selected parts of the manuscript; and H.Y.E., A.B., R.M., and C.I.E.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: C. I. Edvard Smith, Department of Laboratory Medicine, Clinical Research Center, Karolinska Institutet, Hälsovägen 7, 141 86 Huddinge, Stockholm, Sweden; e-mail: edvard.smith@ki.se; or Robert Månsson, Department of Laboratory Medicine, Center for Hematology and Regenerative Medicine, Karolinska Institutet, Blickagången 16, 141 83 Huddinge, Stockholm, Sweden; e-mail: robert.mansson@ki.se.