

Key Points
B-ALL subgroups with extramedullary disease, TP53 mutation, BCR-ABL+, or posttransplant relapse can benefit from CD19 CAR T-cell therapy.
CAR T-cell therapy followed by subsequent consolidative allo-HSCT showed better LFS and OS than CAR T-cell therapy alone.

Abstract
Anti-CD19 chimeric antigen receptor (CAR) T-cell therapy is effective in patients with advanced B-cell acute lymphoblastic leukemia (B-ALL). However, efficacy data is sparse in subgroups of patients with high-risk features such as BCR-ABL+, TP53 mutation, extramedullary disease (including central nervous system leukemia) or posttransplant relapse. It is also uncertain whether there is an added benefit of transplantation after anti-CD19 CAR T-cell therapy. We conducted a phase 1/2 study of 115 enrolled patients with CD19+ B-ALL. A total of 110 patients were successfully infused with anti-CD19 CAR T cells. In all, 93% of patients achieved a morphologic complete remission, and 87% became negative for minimal residual disease. Efficacy was seen across all subgroups. One-year leukemia-free survival (LFS) was 58%, and 1-year overall survival (OS) was 64% for the 110 patients. Seventy-five nonrandomly selected patients (73.5%) subsequently received an allogeneic hematopoietic stem cell transplant (allo-HSCT). LFS (76.9% vs 11.6%; P < .0001; 95% confidence interval [CI], 11.6-108.4) and OS (79.1% vs 32.0%; P < .0001; 95% CI, 0.02-0.22) were significantly better among patients who subsequently received allo-HSCT compared with those receiving CAR T-cell therapy alone. This was confirmed in multivariable analyses (hazard ratio, 16.546; 95% CI, 5.499-49.786). Another variate that correlated with worse outcomes was TP53 mutation (hazard ratio, 0.235; 95% CI, 0.089-0.619). There were no differences in complete remission rate, OS, or LFS between groups of patients age 2 to 14 years or age older than 14 years. Most patients had only mild cytokine release syndrome and neurotoxicity. Our data indicate that anti-CD19 CAR T-cell therapy is safe and effective in all B-ALL subgroups that have high-risk features. The benefit of a subsequent allo-HSCT requires confirmation because of nonrandom allocation. This trial was registered at www.clinicaltrials.gov as #NCT03173417.
Introduction
Relapsed or refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL) is associated with extremely poor prognosis and remains a leading cause of death for pediatric and young adult leukemia patients.1-4 The development of anti-CD19 chimeric antigen receptor (CAR) T-cell therapy has been a milestone for these patients. Since 2011, several large clinical trials of anti-CD19 CAR T-cell therapy have demonstrated excellent efficacy for patients with R/R B-ALL. With complete remission (CR) rates as high as 68% to 93%, it is now possible to offer a cure for some of these patients.5-10 However, relapse remains common over time and occurs in ∼40% to 50% of patients.5,11,12 Furthermore, there are still questions regarding which patients might benefit and whether there are subgroups of patients whose response to this novel therapy is better or worse survival. We conducted a phase 1/2 single-center study using anti-CD19 CAR T cells to treat patients with R/R B-ALL, including those with high-risk features such as BCR-ABL fusion gene, TP53 mutation, extramedullary disease (EMD) (including central nervous system [CNS] leukemia), and those who relapsed after allogeneic hematopoietic stem cell transplantation (allo-HSCT).
Methods
Trial design and participants
The primary objective of this phase 1/2 study was to assess the efficacy and safety of anti-CD19 CAR T cells in patients with R/R B-ALL, including subgroups with high-risk features. The study was approved by the Lu Daopei Hospital Ethics Committee and was conducted in accordance with the Declaration of Helsinki; all patients provided informed consent.
Clinical procedures
Patients with CD19+ R/R B-ALL between ages 2 and 75 years with an Eastern Cooperative Oncology Group score between 0 and 3 were eligible. Inclusion and exclusion criteria are detailed in supplemental Methods. Patients with EMD or previous allo-HSCT without active graft-versus-host disease (GVHD) were eligible. Patients with diffuse EMD were confirmed via biopsy and fluorodeoxyglucose-avid positron emission tomography/computed tomography scans. Patients with CNS leukemic involvement were also eligible for the study, provided they were asymptomatic. Patients with significant neurologic deterioration were not eligible until alternative therapies achieved neurologic stabilization and the patient’s status returned to baseline.13 CNS disease status was defined as CNS-1 (no detectable blasts in white blood cell counts in a sample of cerebrospinal fluid), CNS-2 (blast cells in white blood cell counts detected in a sample with <5 leukocytes per mL and <10 erythrocytes per mL), or CNS-3 (blast cells detected in a sample with ≥5 leukocytes per mL and <10 erythrocytes per mL).7 CR, CR with incomplete count recovery (CRi), and minimal residual disease (MRD) were defined in accordance with the 2018 National Comprehensive Cancer Network guidelines.14 MRD in bone marrow (BM) and peripheral blood (PB) was assessed by flow cytometry (sensitivity 1:10 000). Disease assessment is detailed in supplemental Methods.
All 115 patients received fludarabine (30 mg/m2 per day) and cyclophosphamide (250 mg/m2 per day) lymphodepleting chemotherapy for 3 consecutive days before CAR T-cell infusion (day –5 to day –3). The median time from leukapheresis to CAR T-cell infusion was 14 days (range, 9-35 days). A total of 85 (73.9%) of 115 patients received systemic bridging chemotherapy between leukapheresis and fludarabine-cyclophosphamide lymphodepletion to control rapid disease progression. Seventy-one patients received low- to median-intensity bridging chemotherapy, and the remaining 14 had intensive or count suppressive treatment.
On the basis of our previous study,15 the highest response rate occurred in patients who received a single infusion of CD19 CAR T cells at a dose of ≥1 × 105 CAR T cells per kg. Therefore, in this study, the targeted infusion dose was 1 × 105 to 10 × 105 CAR T cells per kg. Anti-CD19 CAR T cells from 110 of 115 of patients were successfully manufactured and infused in a single dose. At the beginning of this study, there were 21 patients in each group who received CAR T cells with either the 4-1BB or the CD28 costimulatory domain. After confirming that the 4-1BB group had better efficacy and safety (Xiang-Yu Zhao, J.Y., X. Zhang, X.-a.L., Min Xiong, Jian-Ping Zhang, X. S. Zhou, Fei-Fei Qi, T.H., Yan-Ping Ding, Xue-Lian Hu, P.L., and Xiao-Jun Huang, manuscript in preparation), 4-1BB as the costimulatory domain was used after November 2017 for all subsequent 68 enrolled patients (Figure 1).

Once patients had achieved CR, they were given options to proceed or not proceed to consolidative allo-HSCT. For patients bridging into transplantation, conventional myeloablative pretransplantation conditioning regimens were given; a regimen based on total body irradiation for those age older than 5 years or a regimen based on busulfan for those age 5 years or younger. The transplant donor source was an HLA-identical sibling, matched-unrelated, or haploidentical in that order of preference. Cyclosporin A, short-term methotrexate, and mycophenolate mofetil were used for GVHD prophylaxis.
Anti-CD19 CAR T-cell manufacture
CAR T cells were manufactured by using peripheral blood mononuclear cells (PBMCs) collected by leukapheresis. In the 94 patients without a previous transplant, autologous PBMCs were collected. In patients who had relapsed after transplant, 14 of 16 used autologous PBMCs and 2 of 16 used PBMCs derived from their transplant donors.
CAR T cells were produced using the Good Manufacturing Practice facilities at Beijing Immunochina Pharmaceuticals Co., Ltd. The 4-1BB CAR consisted of anti-CD19 scFv FMC63, a CD8α hinge, a TM domain, and 4-1BB and CD3ζ cytoplasmic domains. The CD28 CAR consisted of anti-CD19 scFv FMC63, a CD28 hinge, TM, and the CD3ζ cytoplasmic domain. Briefly, the PBMCs from each patient were prepared using Ficoll (GE Healthcare, Boston, MA) density centrifugation leukapheresis. The PBMCs were washed twice in saline, and T cells were isolated and activated using CD3/CD28 magnetic beads (Thermo Fisher, Waltham, MA) at a T-cell:bead ratio of 1:1. T cells were cultured in X-VIVO 15 medium (Lonza Group, Basel, Switzerland) supplemented with 500 U/mL of interleukin-2 at a density of 1.5 × 106 cells per mL. Cells were transduced with Good Manufacturing Practice–grade lentiviral vector carrying the CAR components. Cell viability and transduction efficiency were monitored for 5 to 7 days after lentivirus transduction. When the CAR T cells were sufficiently expanded for patient infusion, the cells were cryopreserved, and they underwent quality control evaluations for viability, potency, CAR expression ratio, replication-competent lentivirus, sterility, mycoplasma contamination, and endotoxin levels. All CAR T-cell products met the defined specifications. Assessment of response after CAR T-cell treatment
The primary end points were to evaluate short-term efficacy and safety, including CR, and MRD-negative CR on day 30, and CAR T-cell–related cytokine release syndrome (CRS) and neurotoxicity in patients with R/R B-ALL, including subgroups with high-risk features. The secondary end points were to assess 1-year overall survival (OS) and leukemia-free survival (LFS) after CAR T-cell therapy. OS was calculated from the date of CAR T-cell infusion to the date of last follow-up or death. LFS was calculated from the date of CR to the date of relapse, death, or the last follow-up.
Assessment of toxicities after CAR T-cell treatment
CRS and neurotoxicity were graded according to American Society for Transplantation and Cellular Therapy consensus guidelines.16 The CRS was considered to be severe if it was grade 3 or higher. Severe neurotoxicity was defined as a seizure of any grade or toxicity of grade 3 or higher.
Statistical analysis
The probabilities of OS and LFS were estimated by using the Kaplan-Meier method and were compared with the log-rank test by using GraphPad Prism 5 software. CR rates of subgroups, as well as CAR peak and toxicity effects, including CRS and neurotoxicity, were estimated by crosstab descriptive statistics constructed by SPSS software (version 16.0) and were compared using χ2 tests between different subgroups of patients. Forward selection was used to select appropriate variables for multivariable Cox proportional hazards regression models for OS and LFS of subgroups, CR rate, CRS, and neurotoxicity. P < .05 was considered statistically significant. All the reported P values are 2-sided.
Results
From 1 April 2017 to 1 May 2018, a total of 115 patients with R/R B-ALL were consecutively enrolled. In all, 110 patients ranging from 2 to 61 years of age (71 patients were between age 2 and 14 years) were successfully infused with anti-CD19 CAR T cells. Patients’ characteristics are summarized in Tables 1 and 2.
Characterizations of anti-CD19 CAR T-cell products
Patients were divided into 2 dosing groups with the target CAR T-cell dose of either ≥1 × 105 to <3 × 105 cells per kg or ≥3 × 105 to ≤10 × 105 cells per kg. The median dose of infused cells was 3 × 105 (range, 0.2-10 × 105) CAR T cells per kg. Although 5 patients received less than the target dose of 1 × 105 CAR T cells per kg because of inadequate manufacture, all achieved CR on day 30. The median CAR T-cell transfection efficiency was 51.4% (range, 8.7%-90.2%), and the median CAR T-cell percentage peak of total lymphocytes in PB was 23.9% (range, 0.1%-90.6%) appearing at days 7 to 11 after infusion. CAR T-cell expansion was significantly higher when BM blasts were ≥5% vs <5% (CAR T-cell peak, 34.2 ± 2.9 vs 19.6 ± 2.8; P = .001), or when the costimulatory domain was 4-1BB vs CD28 (CAR T-cell peak, 31.3 ± 2.6 vs 17.4 ± 3.0; P = .013), but it was significantly lower in the presence of EMD (CAR T-cell peak, 17.7 ± 3.0 vs 31.6 ± 2.6; P = .010) (Table 3).
Efficacy on day 30 after CAR T-cell treatment
A total of 102 patients (92.7%) achieved morphologic CR at 30 days after anti-CD19 CAR T-cell infusion with 60% CRi. Moreover, 96 (87.3%) of 110 patients had an MRD-negative CR. Six patients were MRD-positive in the cohort of 102 patients with CR. Seventy-one pediatric patients achieved an initial high CR rate after CAR T-cell therapy, regardless of their previous response disease status before CAR T-cell therapy (Table 2).
Table 3 provides the CR rates for the subgroups. No difference in CR rates was observed between the 2 groups receiving different CAR T-cell doses. Fourteen patients had BCR-ABL+ B-ALL, including 3 patients with blast crisis from chronic myeloid leukemia. Ten patients had BCR-ABL P190 and 4 had BCR-ABL P210. On day 30, all patients who had achieved morphologic CR showed no differences compared with BCR-ABL– B-ALL patients. However, 12 (86%) of 14 patients had MRD-negative CR as judged from flow cytometry; 5 (36%) of 14 achieved BCR-ABL− CR on day 30, which later increased to 9 (64%) of 14 by day 60. There were 12 patients with TP53 mutation. Morphologic CR rate was 91.7% (11 of 12). There were no significant differences in CR rates between the TP53+ mutation and TP53– mutation groups.
Twenty-three patients had EMD with median circulating PB blasts at 6.5% (range, 0% to 30%) vs 0% (range, 0% to 65%) in the 87 patients without EMD. There was no statistically significant difference between the 2 groups. Of the 17 patients with CNS leukemia, 4 had CNS-3 disease. Only 1 patient had CNS leukemia without BM blasts. The median blast counts in the cerebrospinal fluid by flow cytometry were 414 cells per mL (range, 20-8415). The CR rate in patients with CNS leukemia was lower than the rate in those without CNS leukemia (76.5% vs 95.7%; P = .021). MRD-negative CR rate was also lower for patients with CNS leukemia (among the total of 6 patients who had MRD-positive disease, 3 had CNS leukemia).
Forty-four patients had BM blasts >20%. Lower CR rate was achieved in this group compared with the group that had BM blasts ≤20% (84.1% vs 98.5%; P = .015). The CR rate for patients with BM blasts ≥5% was lower than that in patients with BM blasts <5% (88.2% vs 100%; P = .018).
Sixteen patients had previous allo-HSCT before CAR T-cell therapy, and 11 (68.8%) had at least 1 donor lymphocyte infusion. No patients had active GVHD before CAR T-cell therapy. After CAR T-cell infusion, 15 (93.8%) of 16 of patients achieved CR. No statistically significant difference was observed in the CR rate after CAR T-cell treatment for patients who had previous allo-HSCT vs those without.
Long-term efficacy after CAR T-cell treatment
The median follow-up time for the entire cohort was 233.5 days (range, 27-478 days). For the total 110 infused patients, 1-year OS was 63.9%, and 1-year LFS was 57.9% (Figure 2A-B). For the 102 patients who achieved CR, 1-year OS was 67.3% and 1-year LFS was 63.2% (Figure 2C-D). The 1-year OS was 79.1% and the 1-year LFS was 76.9% for patients bridging into allo-HSCT after CAR T-cell therapy, whereas the 1-year OS was 32% and the 1-year LFS was 11.6% for patients who received CAR T-cell therapy alone (Figure 3A-B). There was a trend toward better OS (85.5% vs 59.7%; P = .193; 95% CI, 0.739-4.462) and LFS (73.3% vs 57.4%; P = .123; 95% CI, 0.25-1.18) for patients with CR vs CRi (Figure 4A-B). There was also a trend toward better OS (69.7% vs 50%; P = .136; 95% CI, 0.65-22.61) and LFS (64.8% vs 42.9%; P = .135; 95% CI, 0.22-0.92) for patients with MRD-negative CR vs MRD-positive CR (Figure 5A-B), but no significant difference was found because the number of patients in the MRD-positive patient group (n = 6) was too small. A total of 23 (22.5%) of 102 patients subsequently relapsed. Seven patients who had a relapse were CD19– and the remaining 16 patients were CD19+.
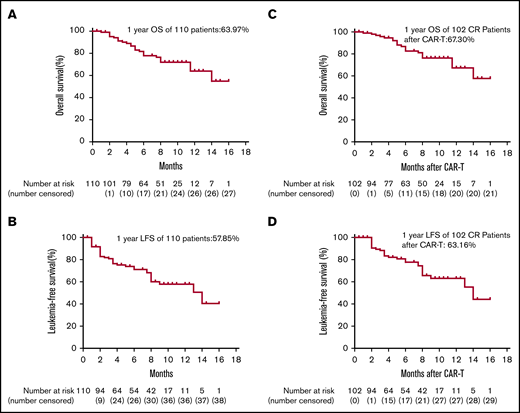
Kaplan-Meier plots of OS and LFS of 110 total patients and 102 CR patients after anti-CD 19 CAR T-cell therapy. (A-D) On day 30 after anti-CD19 CAR T-cell infusion, 102 of 110 patients achieved morphologic CR. To show long-term efficacy after CAR T-cell therapy, Kaplan-Meier analysis of OS and LFS were performed for both the 110 total patients and 102 CR patients.
Kaplan-Meier plots of OS and LFS of 110 total patients and 102 CR patients after anti-CD 19 CAR T-cell therapy. (A-D) On day 30 after anti-CD19 CAR T-cell infusion, 102 of 110 patients achieved morphologic CR. To show long-term efficacy after CAR T-cell therapy, Kaplan-Meier analysis of OS and LFS were performed for both the 110 total patients and 102 CR patients.
![Long-term efficacy of allo-HSCT after CAR T-cell therapy. (A-B) Of the 102 CR patients, 75 nonrandomly selected patients subsequently were bridged into allo-HSCT (designated the "CAR T bridged into allo-HSCT" group). The remaining 27 patients did not undergo transplantation (designated the "CAR T alone" group). Kaplan-Meier analysis showed that 1-year OS and LFS for patients bridging into allo-HSCT after CAR T-cell therapy were significantly better than those for patients receiving CAR T-cell therapy alone (OS: 79.1% vs 32.0%; P < .0001; hazard ratio [HR], 0.048; 95% CI, 0.016-0.148; LFS: 76.9% vs 11.6%; P < .0001; HR, 35.45; 95% CI, 11.6-108.4).](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/10/10.1182_bloodadvances.2020001466/2/m_advancesadv2020001466f3.png?Expires=1764981472&Signature=I-Lcc9d01sAJd5xUYlHkwxpu14yYBsZb~K4BqC8MlUbViOjLAsb5eGep2GzzJa5dxr4HpNeJmoias25PmHtQaA074d71uBEwVCAVN6sr4h0oipm43yYx8oInTpGGZyDYYT1bSYBKyl3hloEdrvl2S-EV~KMaFXTI77HL7rjWU7te2orIhnU9J1uEKV0P6WCe3zZh1X1sbYij0butmSPn19pGGewRStzzdxYuqDNOu5CMcbcN-JpsMBaBJj4zkP1j~pG2mPkwI5OlnW2YDP5mpiLETBVqjKESKU0skfNzwWwoS9peeeSmDw5BIXO~GgEBv6NW4Uhz0HkLkIPClsdAnw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Long-term efficacy of allo-HSCT after CAR T-cell therapy. (A-B) Of the 102 CR patients, 75 nonrandomly selected patients subsequently were bridged into allo-HSCT (designated the "CAR T bridged into allo-HSCT" group). The remaining 27 patients did not undergo transplantation (designated the "CAR T alone" group). Kaplan-Meier analysis showed that 1-year OS and LFS for patients bridging into allo-HSCT after CAR T-cell therapy were significantly better than those for patients receiving CAR T-cell therapy alone (OS: 79.1% vs 32.0%; P < .0001; hazard ratio [HR], 0.048; 95% CI, 0.016-0.148; LFS: 76.9% vs 11.6%; P < .0001; HR, 35.45; 95% CI, 11.6-108.4).
Long-term efficacy of allo-HSCT after CAR T-cell therapy. (A-B) Of the 102 CR patients, 75 nonrandomly selected patients subsequently were bridged into allo-HSCT (designated the "CAR T bridged into allo-HSCT" group). The remaining 27 patients did not undergo transplantation (designated the "CAR T alone" group). Kaplan-Meier analysis showed that 1-year OS and LFS for patients bridging into allo-HSCT after CAR T-cell therapy were significantly better than those for patients receiving CAR T-cell therapy alone (OS: 79.1% vs 32.0%; P < .0001; hazard ratio [HR], 0.048; 95% CI, 0.016-0.148; LFS: 76.9% vs 11.6%; P < .0001; HR, 35.45; 95% CI, 11.6-108.4).
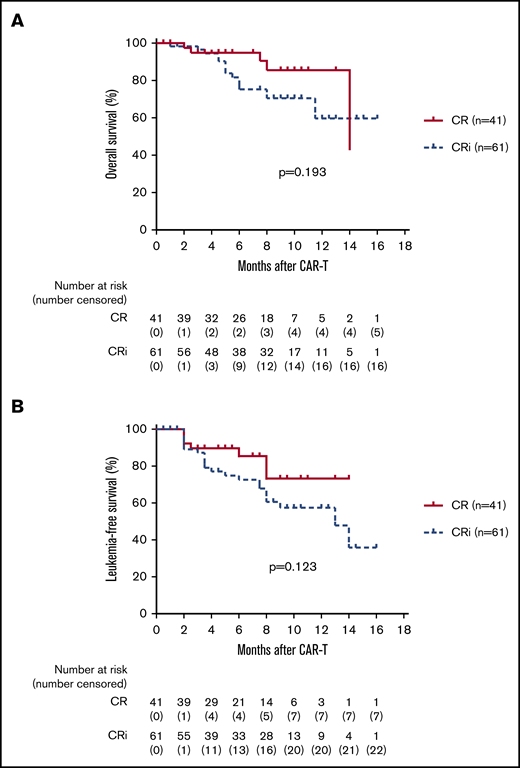
Kaplan-Meier plots of OS and LFS of 102 patients with CR vs CRi. (A-B) On day 30 post anti-CD19 CAR T-cell infusion, a total of 102 patients achieved morphologic CR with 60% (61 of 102) achieving CRi. There was a trend toward better OS (85.5% vs 59.7%; log-rank test P = .193; HR, 1.816; 95% CI, 0.739-4.462) and LFS (73.3% vs 57.4%; P = .123; HR, 0.54; 95% CI, 0.25-1.18) for patients with CR vs CRi.
Kaplan-Meier plots of OS and LFS of 102 patients with CR vs CRi. (A-B) On day 30 post anti-CD19 CAR T-cell infusion, a total of 102 patients achieved morphologic CR with 60% (61 of 102) achieving CRi. There was a trend toward better OS (85.5% vs 59.7%; log-rank test P = .193; HR, 1.816; 95% CI, 0.739-4.462) and LFS (73.3% vs 57.4%; P = .123; HR, 0.54; 95% CI, 0.25-1.18) for patients with CR vs CRi.
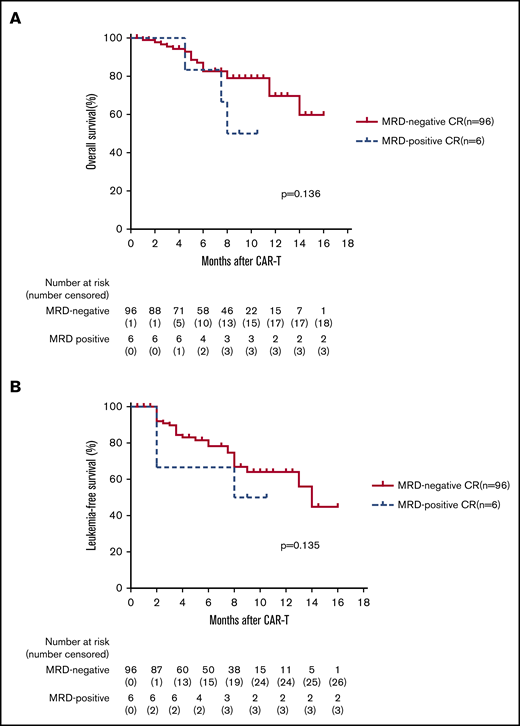
Kaplan-Meier plots of OS and LFS of 102 patients with MRD-negative CR vs MRD-positive CR. (A-B) For the 102 patients who achieved CR after CAR T-cell therapy, 96 patients were MRD-negative CR. Kaplan-Meier analysis demonstrated a trend toward better OS (69.7% vs 50%; P = .136; HR, 3.85; 95% CI, 0.65-22.61) and LFS (64.8% vs 42.9%; P = .135; HR, 1.62; 95% CI, 0.38-7.0) for patients with MRD-negative CR vs MRD-positive CR, but there was no statistical significance because of the small number of patients in the MRD-positive patient group (n = 6).
Kaplan-Meier plots of OS and LFS of 102 patients with MRD-negative CR vs MRD-positive CR. (A-B) For the 102 patients who achieved CR after CAR T-cell therapy, 96 patients were MRD-negative CR. Kaplan-Meier analysis demonstrated a trend toward better OS (69.7% vs 50%; P = .136; HR, 3.85; 95% CI, 0.65-22.61) and LFS (64.8% vs 42.9%; P = .135; HR, 1.62; 95% CI, 0.38-7.0) for patients with MRD-negative CR vs MRD-positive CR, but there was no statistical significance because of the small number of patients in the MRD-positive patient group (n = 6).
Efficacy of CAR T-cell treatment of B-ALL patients with TP53 mutation
The OS and LFS at 6 months were much lower for patients with a TP53 mutation when compared with patients without (OS, 51.9% vs 89.0%; P < .0001; 95% CI, 11.35-336.1; LFS, 42.4% vs 82.6%; P = .0002; 95% CI, 0.013-0.26; Figure 6A-B). Five of 11 patients relapsed, with the median time to relapse of 56 days (range, 40-225 days), including 2 EMD relapses. Seven patients died, including 2 from GVHD after transplantation, 1 from infection, and 4 from relapse. The median time to death was 142 days (range, 63-346 days).
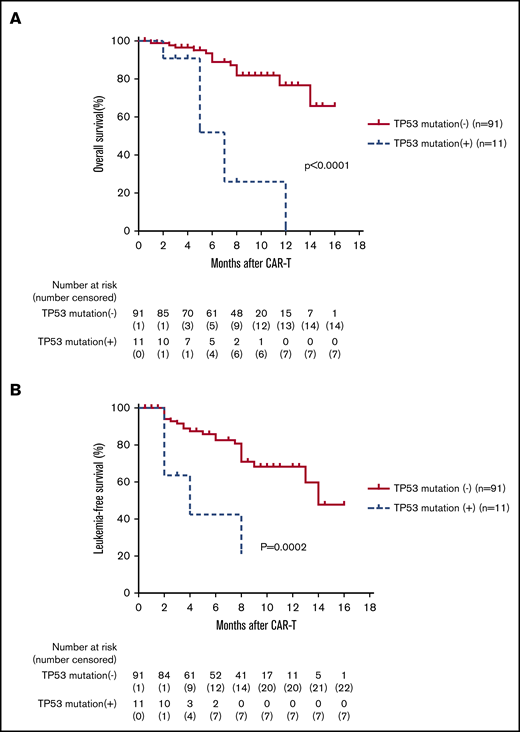
B-ALL patients with TP53 mutation had a lower OS and LFS than those without TP53 mutation. (A-B) The 102 patients were divided into 2 groups based on TP53 mutation status: TP53+ mutation and TP53– mutation. Kaplan-Meier analysis showed that OS and LFS at 6 months were much lower for patients carrying the TP53 mutation when compared with the patients without a TP53 mutation (OS: 51.9% vs 89.0%; P < .0001; HR, 61.75; 95% CI, 11.35-336.1; LFS: 42.4% vs 82.6%; P = .0002; HR, 0.06; 95% CI, 0.014-0.26).
B-ALL patients with TP53 mutation had a lower OS and LFS than those without TP53 mutation. (A-B) The 102 patients were divided into 2 groups based on TP53 mutation status: TP53+ mutation and TP53– mutation. Kaplan-Meier analysis showed that OS and LFS at 6 months were much lower for patients carrying the TP53 mutation when compared with the patients without a TP53 mutation (OS: 51.9% vs 89.0%; P < .0001; HR, 61.75; 95% CI, 11.35-336.1; LFS: 42.4% vs 82.6%; P = .0002; HR, 0.06; 95% CI, 0.014-0.26).
Efficacy of CAR T-cell treatment for patients who relapsed after allo-HSCT
Patients with a history of previous transplantation had a lower 1-year OS and LFS than those without previous transplantation (OS, 30.5% vs 79.2%; P = .009; 95% CI, 1.51-18.48; LFS, 25.4% vs 69.4%; P = .011; 95% CI, 0.07-0.71; Figure 7A-B). After CAR T-cell therapy, 2 patients developed acute GVHD (grade 1 and grade 3), and 2 patients developed extensive chronic GVHD. Six of 15 patients remained in MRD-negative CR at 3 to 16 months. Two patients underwent a second allo-HSCT and remained disease free at 19 months and 4 months after transplant, respectively.
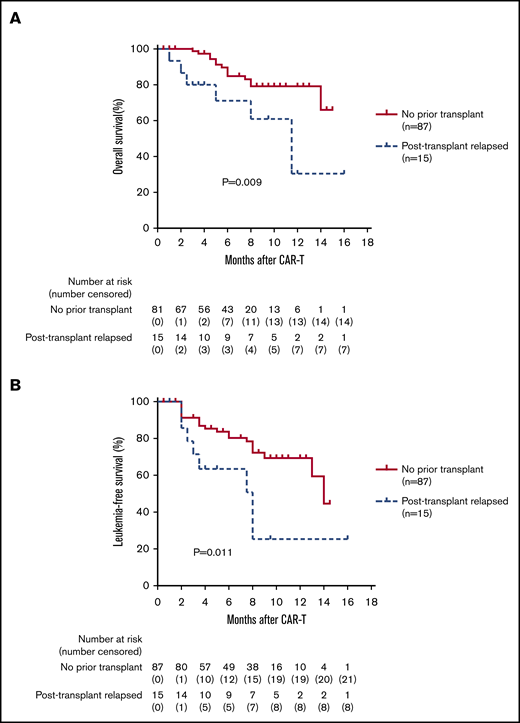
Long-term efficacy of CAR T-cell therapy in patients who relapsed after transplantation. (A-B) The 102 CR patients who received CAR T-cell therapy were divided into 2 groups: those who relapsed after transplant and those without previous transplant. Patients with a history of previous transplantation had a lower 1-year OS and LFS than the group without previous transplant (OS: 30.5% vs 79.2%; HR, 5.27; P = .009; 95% CI, 1.51-18.48; LFS: 25.4% vs 69.4%; P = .011; HR, 0.23; 95% CI, 0.07-0.71).
Long-term efficacy of CAR T-cell therapy in patients who relapsed after transplantation. (A-B) The 102 CR patients who received CAR T-cell therapy were divided into 2 groups: those who relapsed after transplant and those without previous transplant. Patients with a history of previous transplantation had a lower 1-year OS and LFS than the group without previous transplant (OS: 30.5% vs 79.2%; HR, 5.27; P = .009; 95% CI, 1.51-18.48; LFS: 25.4% vs 69.4%; P = .011; HR, 0.23; 95% CI, 0.07-0.71).
Efficacy of CAR T-cell treatment of patients bridging into allo-HSCT vs those who did not bridge into allo-HSCT
Once patients achieved CR after CAR T-cell treatment, they were given options to proceed to consolidative allo-HSCT or not. Of the 102 CR patients, 75 (69 MRD negative, 6 MRD positive) nonrandomly selected patients subsequently underwent transplantation with a median time of 63 days (range, 36-120 days) after CAR T-cell infusion. Characteristics of patients who bridged into transplantation vs those who did not are provided in Table 4. A total of 10 (13.3%) of 75 patients who received a transplant relapsed (8 BM and 2 EMD relapses) with a median relapse time of 225 days (range, 60-385 days); 7 (10.1%) of 69 MRD-negative CR patients relapsed vs 3 (50%) of 6 MRD-positive CR patients. There were 5 deaths in this group, including 1 from septic shock, 2 from GVHD, and 2 from relapse.
For the remaining 27 patients who received CAR T-cell therapy alone, 13 patients relapsed after previous allo-HSCT. After CAR T-cell therapy, 4 of 13 patients relapsed within 3 months, which precluded them from a second transplant, and 3 of 13 died within 3 months. The remaining 6 of 13 patients did not bridge into a second transplantation for reasons such as personal preference, performance status, financial issues, and donor availability.
Among the remaining 14 patients without a previous transplant, 6 had early relapse in 3 months, 1 had a follow-up time of less than 3 months, and 7 did not undergo consolidative transplantation for personal preference or financial reasons. A total of 13 (48.2%) of 27 patients relapsed with a median relapse time of 100 days (range, 52-248 days). Seven (55.6%) had CD19+ relapse and 6 (44.4%) had CD19– relapse. The 1-year OS and LFS for patients bridging into allo-HSCT after CAR T-cell therapy were significantly better than those for patients receiving CAR T-cell therapy alone (OS, 79.1% vs 32.0%; P < .0001; 95% CI, 0.02-0.22; LFS, 76.9% vs 11.6%; P < .0001; 95% CI, 11.6-108.4; Figure 3A-B).
Efficacy for other subgroups
No significant differences in OS and LFS were observed for patients age 2 to 14 years vs patients age older than 14 years, or those who received a CAR T-cell dose <3 × 105 vs ≥3 × 105 cells per kg. There were also no differences in OS and LFS for patients with EMD vs those without, BM blasts >20% vs ≤20%, BM blasts <5% vs ≥5%, or BCR-ABL+ vs BCR-ABL– groups (supplemental Figure S2). For the BCR-ABL+ group, 3 (21.4%) of 14 patients subsequently relapsed with a median time of 60 days to relapse (range, 56-274 days).
Adverse effects
CRS occurred in 102 (92%) of 110 patients; 84 (76%) of the 110 patients had grade 1 to 2 CRS and 18 (16%) of 110 had grade 3 to 4 CRS. Fourteen patients had grade 3 CRS, and 4 patients had grade 4 CRS. Dexamethasone was given to 14 patients and tocilizumab to 10 patients with grade 3 to 4 CRS. The main adverse effect was fever (supplemental Figure S3).
Of the 110 patients, 23 (21%) had neurotoxicity, 15 (14%) had grade 2 to 3 neurotoxicity, and 8 (7%) had grade 1 neurotoxicity.17-19 Neurotoxicity was managed according to Gust et al.18 To manage seizure (13 of 110) and loss of consciousness, anticonvulsants such as diazepam, phenobarbital, and dexamethasone were given accordingly. In the 17 patients with CNS leukemia, 3 had neurotoxicity. One patient with CNS-3 status developed grade 1 neurotoxicity, and 2 patients with CNS-1 status had grade 1 or 3 neurotoxicity, respectively.
We found that BM blasts ≥5%, BM blasts >20%, and CAR T-cell therapy with CD28 costimulatory domain were significantly associated with higher incidence of grade 3 to 4 CRS and more severe neurotoxicity. Patients who received ≥3 × 105cells per kg had a tendency toward higher incidence of grade 3 to 4 CRS and severe neurotoxicity, but they were not statistically different than those who received <3 × 105 cells per kg (Table 3).
Multivariable analysis
In multivariable analysis, the presence of a TP53 mutation was associated with inferior OS and LFS. Bridging into allo-HSCT after CAR T-cell therapy was associated with improved OS and LFS (Table 5). The group with BM blasts ≤20% had a higher OS than the group with BM blasts >20%; however, there was no difference in LFS. The presence of EMD or posttransplant relapse was not predictive of OS or LFS.
Discussion
Our study addresses the efficacy and safety of the anti-CD19 CAR T-cell treatment in a large cohort of patients with R/R B-ALL with high-risk features at a single institution, which has afforded a better understanding of the nature and the durability of patient response to the treatment.
Our data demonstrated that CR was achieved in 93% of patients 30 days after CAR T-cell infusion. Most importantly, 87% of patients achieved MRD-negative CR. One-year OS was 67.3% and 1-year LFS was 63.2% for the 102 CR patients. Thus, all patients with B-ALL in each high-risk subgroup of BCR-ABL+, TP53 mutation, EMD, CNS leukemia, or posttransplant relapse benefited from the CAR T-cell treatment with 87% to 100% CR rate. Age was not a predictor for treatment outcome. There were no differences in the rates of CR, OS, or LFS for patients in groups with age 2 to 14 years or older than age 14 years.
Patients with BM blasts ≤20% achieved a higher CR rate than those with BM blasts >20% (98.5% vs 84.1%; P = .015). However, there were no differences in LFS. We think this is likely because bridging patients with BM blasts >20% into allo-HSCT helped improve their prognosis.
Twenty-three patients with EMD had a CR rate of 86.9%. Of the 23 patients, 17 had CNS leukemia, including 4 with CNS-3 status. Whether patients with CNS leukemia can be safely and effectively treated with CAR T-cell therapy has been controversial.6,18-21 In our study, the CR rate of patients with CNS leukemia was lower than that of patients without CNS leukemia (76.5% vs 95.7%; P = .021), but there was no difference in OS or LFS.
Published data on the efficacy of anti-CD19 CAR T-cell therapy in eradicating BCR-ABL leukemia in BCR-ABL+ B-ALL have been conflicting.5,22,23 In our cohort, 14 patients with BCR-ABL+ translocation, including 3 that were transformed from chronic myeloid leukemia achieved a CR rate of 100% by day 30. Only 36% achieved negative BCR-ABL status; the percentage later increased to 69% on day 60. This suggests that it takes time for CAR T-cell therapy to eliminate leukemia cells to achieve negative BCR-ABL status. Importantly, no significant differences were observed in OS and LFS between BCR-ABL+ and BCR-ABL– groups.
TP53 mutation or deletion in B-ALL is a poor prognostic indicator in both adults and pediatric patients.24-26 Although the CR rate (91.7%) was high in 12 patients with TP53 mutation, half of them relapsed quickly with a median relapse time of 56 days. The multivariable analysis showed that TP53 mutation status was the major independent indicator of poor prognosis even after CAR T-cell treatment. The underlying reason remains unknown. Given the initial high CR rate on day 30, it is less likely that poor prognosis is a result of decreased susceptibility of leukemia cells to CAR T-cell therapy. TP53 mutations accelerate the genomic instability and evolution of cancer cells and support their growth and survival.24,26,27 The higher rate and earlier onset of relapse in patients with TP53 mutation could be a result of accelerated mutations in or increased proliferation or survival capacity of leukemic blasts and/or evasion of CAR T-cell cytotoxicity. With early relapse and poor prognosis, consolidative allo-HSCT within 2 months should be considered.
Once patients relapse after allo-HSCT, currently available treatments are often unsatisfactory.28 However, CAR T-cell therapy now offers some hope.6,7 In our study, the CR rate after CAR T-cell therapy was 93.8% (15 of 16) for the 16 patients who relapsed after previous allo-HSCT. However, 1-year OS and LFS for patients with previous HSCT were much worse than those without. Only 2 patients underwent a second allo-HSCT after CAR T-cell treatment. Previous allo-HSCT is a risk factor, because a majority of patients in this group did not bridge into a second transplant for various reasons.
Despite the initial high CR from CAR T-cell treatment of R/R B-ALL, relapse is still a major problem, with reported relapse rates of 41% to 45%,5,11,12 especially when the tumor burden is high.5 There is much controversy about whether transplantation is necessary after CAR T-cell therapy.5-7,11 Park et al5 from the Memorial Sloan Kettering Cancer Center found no difference in LFS and OS between patients who underwent allo-HSCT after CAR T-cell therapy and those who did not. In their study, of the 16 patients who underwent transplantation, 6 subsequently relapsed and another 6 died of transplant-related mortality (TRM). Other studies showed that bridging to HSCT after CAR T-cell therapy could improve LFS.6,7,29 However, most studies had only a small number of patients. We previously observed that bridging into allo-HSCT after CAR T-cell therapy for R/R B-ALL improves LFS and OS.15,30,31 In this large cohort study, we again demonstrated that only 10 (13.3%) of 75 patients in the CAR T-cell therapy group that bridged into the transplant group relapsed whereas 13 (48.2%) of 27 patients who received CAR T-cell therapy alone relapsed. Of the patients who underwent CAR T-cell therapy and allo-HSCT, the 1-year OS was 79.1%, and LFS was 76.6%. Multivariable analyses confirmed that patients who bridged to allo-HSCT after CAR T-cell therapy had better OS and LFS. We speculate that haploidentical transplantation (67%) in the majority of our patients and a myeloablative conditioning regimen in all of our patients may have played a role in decreasing leukemia relapse. There were more patients with high-risk features in our study who may benefit more from transplantation. Overall, our TRM rate was low. Only 3 of 75 patients died as a result of TRM during the 1-year observation period.
For the 27 patients who did not bridge into allo-HSCT, the 1-year OS was 32.0% and 1-year LFS was 11.6%. Possible explanations for the shorter duration of remission for these patients might be their high-risk leukemia features or might be the result of a lower dose of infused CAR T cells, which is much less likely. The dosage of infused anti-CD19 CAR T cells varied in many B-ALL CAR T-cell trials.7,11 The majority exceeded 1 × 106 cells per kg.6,18 Hay et al29 from the Fred Hutchinson Cancer Research Center reported no difference in LFS for patients receiving 2 × 105 vs 2 × 106 CAR T cells per kg; 49% of the patients relapsed at a median time of 3.5 months. It should also be noted that even with much higher doses of CAR T cells used in other clinical trials, the 1-year relapse rate remained as high as 50%.7 We have previously demonstrated that a lower CAR T-cell infusion dose could achieve equally high CR rates and, at the same time, reduce the risk of severe CRS.15 Here again we showed that our 2 CAR T-cell dose groups of <3 × 105 vs ≥3 × 105 cells per kg could achieve similar high CR rates, LFS, and OS. With a median relatively low dose of 3 × 105 cells per kg, our CRS grade 3 to 4 rate was only 17%, lower than that in most clinical trials that had CRS grade 3 to 4 rates as high as 26% to 88%.5,12 The same was true for neurotoxicity.
We found no statistically significant difference in CRS between the age 2 to 14 years and the age older than 14 years groups. It was suggested that the blood-brain barrier in a child is not well developed and its permeability is high. Consequently, CAR T cells might cross the blood-brain barrier easily and trigger the release of a cascade of cytokines.17 In this study, we found no significant difference in neurotoxicity between the 2 age groups. No difference was observed in the incidence and severity of neurotoxicity in patients with or without CNS leukemia, suggesting that CNS leukemia is not an absolute contraindication for CAR T-cell treatment.
Our findings support the notion that the leukemia burden and the peak in vivo CAR T-cell levels achieved after initial expansion are closely related to the CR rate, CRS, and neurotoxicity. Furthermore, our study indicates that patients treated with CD28 CAR T cells had higher CRS and neurotoxicity compared with those treated with 4-1BB CAR T cells (CRS, 38.1% vs 11.2%; P = .003; neurotoxicity, 33.3% vs 9%; P < .001).
In conclusion, a high CR rate can be achieved for patients with R/R B-ALL who are treated with anti-CD19 CAR T-cell therapy, including patients with high-risk features such as EMD, BCR-ABL+, CNS leukemia, TP53 mutation, and relapse after allo-HSCT. The relapse rate for subgroups with high-risk features after CAR T-cell treatment alone remains high, and CAR T-cell therapy followed by subsequent consolidative allo-HSCT has showed better LFS and OS in our study. However, the benefit of a subsequent allo-HSCT transplant requires confirmation with randomized allocation.
Please e-mail the corresponding author at peihua_lu@126.com for renewable materials, protocols, or data.
Acknowledgments
We thank the courageous patients who participated in this trial and their families, the staff in the laboratory for immunotherapy from Lu Daopei Hospital who performed the CAR T-cell–related tests and cytokine analyses, the physicians in the HSCT department for providing clinical consultation and care, and all the nurses for their devotion and patient care. We also thank the Immunochina Pharmaceuticals Co., Ltd., which provided the CAR T-cell products.
This study was partly funded by Immunochina Pharmaceuticals Co., Ltd.
Authorship
Contribution: P.L., X. Zhang, and J.Y. designed the research; G.Z., L.S., Y. Su, Y. Shi, M.Z., J.H., D.S., F.L., W.L., and Y.W. performed the research; X.-a.L. and T.H. designed the CAR T-cell structure; H.W., H.L., and X. Zhou performed the flow cytometry, cytogenetic, and molecular mutation studies; X. Zhang, W.L., and J.L. analyzed data; and P.L., X. Zhang, and J.L. wrote and revised the article.
Conflict-of-interest disclosure: T.H. and X.-a.-L. are employees of Immunochina Pharmaceuticals Co., Ltd. The remaining authors declare no competing financial interests.
Correspondence: Peihua Lu, Lu Daopei Institute of Hematology, No.22 Tongji South Rd, Yizhuang Economic and Technological Development Zone, Daxing District, Beijing 100176, China; e-mail: peihua_lu@126.com.



