Key Points
Recurrent infused iPSC-derived MKs or platelets expressing FVIII are an effective hemostatic strategy for hemophilia A.
Platelets expressing FVIII are additive with recombinant activated FVII in the setting of hemophilia A.

Abstract
B-domainless factor VIII (FVIII) ectopically expressed in megakaryocytes (MKs) is stored in α granules of platelets (pFVIII) and is capable of restoring hemostasis in FVIIInull mice, even in the presence of circulating inhibitors. However, our prior studies have shown that this ectopically expressed pFVIII can injure developing MKs. Moreover, the known risks of prolonged thrombocytopenia after bone marrow transplantation are significant challenges to the use of this strategy to treat individuals with severe hemophilia A and particularly those with intractable clinically relevant inhibitors. Because of these limitations, we now propose the alternative therapeutic pFVIII strategy of infusing pFVIII-expressing MKs or platelets derived from induced pluripotent stem cells (iPSCs). pFVIII-expressing iPSC-derived MKs, termed iMKs, release platelets that can contribute to improved hemostasis in problematic inhibitor patients with hemophilia A. As proof of principle, we demonstrate that hemostasis can be achieved in vitro and in vivo with pFVIII-expressing platelets and show prolonged efficacy. Notably, pFVIII-expressing platelets are also effective in the presence of inhibitors, and their effect was enhanced with recombinant FVIIa. Human pFVIII-expressing iMKs improved hemostasis in vitro, and derived platelets from infused human pFVIII-expressing iMKs improved hemostasis in FVIIInull mice. These studies indicate the potential therapeutic use of recurrent pFVIII-expressing MK or platelet infusions with prolonged hemostatic coverage that may be additive with bypassing agents in hemophilia A patients with neutralizing inhibitors.
Introduction
Ideal treatment of patients with hemophilia A would involve correction of plasma factor VIII (FVIII) levels by establishing FVIII expression in tissues that physiologically express this protein. Progress in achieving such liver FVIII replacement by gene therapy has been reported.1 Although these therapies target the correct organ, they may not target the natural site of FVIII synthesis (ie, liver sinusoidal endothelial cells),2,3 but otherwise, this gene therapy strategy is likely to be near ideal for a majority of patients with significant hemophilia A. However, in patients with severe hemophilia A and intractable inhibitors, a clinical complication seen in up to 30% of patients,4,5 this near-ideal therapy may be problematic in the absence of concurrent elimination of the inhibitors.6,7
Therapy for bleeds in patients with hemophilia A and clinically relevant inhibitors has been vastly improved recently with the introduction of emicizumab (Hemlibra), a heterodimeric antibody that crossbinds FIX and FX8-10 ; however, breakthrough bleeds still occur with this therapy, and concurrent treatment with other bypassing agents, such as recombinant activated FVII (rFVIIa) or FVIII inhibitor bypass activity, may have significant prothrombotic risk.11,12 Platelet-delivered FVIII (pFVIII) is an alternative concurrent therapy that offers the ability of FVIII to be partially protected from circulating inhibitors. However, assessing the degree of that protection is difficult, even in animal models, such as the murine tail-clip exsanguination assay, which tends to be especially sensitive to low levels of pFVIII.3,13,14 Released pFVIII from α granules is not temporarily or spatially available as plasma FVIII, which may explain its efficacy in the presence of inhibitors and its greater efficacy in some hemostatic models over others.15-18 These differences limit the degree to which pFVIII studies in murine models of hemostasis can be directly translated to medically relevant bleeds seen in patients, including joint, retroperitoneal, and intracranial bleeds.19 Moreover, achieving high levels of pFVIII per platelet has been problematic in mice20 with concurrently reported platelet counts, because ectopically expressed FVIII can cause injury to the expressing megakaryocytes (MKs) as a result of poor intracellular FVIII processing.17,21 The differences between plasma and pFVIII therefore raise concern regarding expressing pFVIII by bone marrow transplantation (BMT) in patients with hemophilia A and inhibitors. (1) High levels of pFVIII expression may injure developing MKs and worsen post-BMT thrombocytopenia. A combination of thrombocytopenia and FVIII deficiency with inhibitors may be clinically challenging, even with the availability of emicizumab. (2) Efficacy of pFVIII in target organs of patients with hemophilia A has not been established. (3) Whether pFVIII may be effective in these target organs in the presence of inhibitors has yet to be tested, despite the existence of an FVIIInull dog model of pFVIII.22-24
We propose an alternative strategy for pFVIII therapy: expressing pFVIII in in vitro–grown MKs and using the pFVIII MKs to generate FVIII-containing platelets for acute or prophylactic care in patients with hemophilia A and clinically relevant inhibitors. In this study, we provide proof of principle for such a strategy, beginning with infused pFVIII-expressing murine MK platelets to simulate in vitro–generated clinical platelets25 and followed by infused pFVIII-expressing human MKs, which we have shown can release platelets upon infusion.26 We show that both pFVIII platelets and MKs improved hemostasis in a rotational thromboelastography assay (ROTEM)20 and in FVIIInull mice.27,28 The effect of such platelets persisted for >72 hours. This therapeutic strategy is effective in the presence of inhibitors and can be additive with rFVIIa. Potential uses and limitations of this infused pFVIII approach are discussed.
Materials and methods
Mice lines and isolation of blood
FVIIInull mice with exon 16 disruption in the F8 gene,25,26 the murine transgenic line h38 expressing human BDFVIII (hBDFVIII) in their platelets,29 and the transgenic hαIIb-expressing line30 have been previously described. Line h38 mice are on the FVIIInull background but are designated simply as line h38. All mice had been previously crossed onto a C57Bl6 background for >10 generations. In all studies, littermate mice 6 to 10 weeks of age were studied. Whole blood of FVIIInull, line h38, and hαIIb-expressing mice were collected from the inferior vena cava in 3.8% sodium citrate.31 Blood was used directly in ROTEM or in vivo infusion studies by isolation of platelets using differential centrifugation.32 Total blood counts were determined using a HemaVet counter (Triad Associates) before infusion. Whole-blood and platelet samples were used within 1 hour of isolation. In some studies, line h38 whole blood was mixed with that of FVIIInull mice to obtain decreased percentages of line h38 platelets.
ROTEM studies with murine blood
Whole murine blood was recalcified to 10 mM with 0.2 M of calcium chloride and then transferred to 37°C ROTEM minicups (Werfern) for assessment via ROTEM thromboelastometry using an INTEM-based assay, as previously described.32,33 INTEM reagent (Werfern), an intrinsic pathway activator of kaolin,34 was used to activate mouse whole blood. Supplemental rFVIIa (final concentration, 1-50 nM; NovoNordisk) was added in some studies along with induced pluripotent stem cell (iPSC)–derived MKs (iMKs). In other studies, human full-length rFVIII (final concentration, 0.01-1.0 ng/mL; Advate; Shire) was added to FVIIInull whole blood as a positive control. The thromboelastometry assay was carried out in 30 minutes, and data were analyzed via ROTEM software (Werfern).
In vivo studies using FVIIInull mice and platelets from hαIIb and h38 mice
To define platelet half-life and total percentage of infused circulating platelets postinfusion, 4 × 108 hαIIb-expressing or calcein AM–loaded platelets were infused into FVIIInull mice and analyzed over 72 hours via flow cytometry.26 Platelets from line h38 mice were infused into FVIIInull mice via tail vein for 2 separate in vivo hemostatic assays. The first was a tail-clip exsanguination assay, which we performed in FVIIInull mice that received up to 4 × 108 line h38 platelets in 100 µL of phosphate-buffered saline (PBS) 0.5 to 72 hours before the injury. Tail clipping was 1.67 mm in diameter, performed using a Persona double-edge safety razorblade.29 The primary end point was the number of mice surviving overnight. The second was the iron chloride (FeCl3)–induced carotid artery injury assay, which we have previously described and shown to correlate with the amount of recombinant hFVIII infused.27 Six- to 8-week-old littermate mice were studied. Both male and female recipient FVIIInull mice were used in these studies, but there was no difference between sex in the area under the curve when infused with 2 × 108 to 4 × 108 line h38 platelets 0.5 hours before the study (supplemental Figure 1).
In other studies, 2 × 108 to 4 × 108 line h38 platelets were infused 0.5 to 72 hours before mice were anesthetized using pentobarbital (80 mg/kg; Akorn Pharmaceuticals); their right carotid artery was then exposed to an FeCl3-saturated 1 × 2 mm Whatman 1 filter for 3 minutes at a 20% weight/volume ratio. Blood flow was measured using a Doppler flow probe (Model 0.5VB; Transonic Systems). Total flow was recorded for 30 minutes, with the volume of blood flow over that timeframe as the primary end point. In some FeCl3 carotid artery injury studies, 0.1 to 2.0 mg of inhibitors per kilogram of mice in a total of 50 µL of water were infused. This cocktail of 2 FVIII inhibitors consisting of 1 µg of ESH8 (BioMedica Diagnostics) per 5 µg of GMA-8021 (Green Mountain Antibodies) was previously used to demonstrate the hemostatic efficacy of pFVIII in line h38 mice.15 The inhibitor was infused via the jugular vein of mice before the FeCl3 carotid injury assay. In some of these studies, rFVIIa was infused immediately before the FeCl3 carotid injury assay as well.
Establishing pFVIII-expressing iMKs
Previously described self-inactivating lentiviral constructs for hBDFVIII, hBDFVIIIR1645H (hBDFVIIIRH), and eukaryotic green fluorescent protein (eGFP), each driven by the murine Cxcl4 proximal promoter, were used in these studies.16,17,29 Infectious viral stocks were produced through transient infection of human embryonic kidney 293T cells with the cytomegalovirus Δ8.2 packaging vector,35 the vesicular stomatitis virus glycoprotein envelope vector,35 and the appropriate lentiviral plasmid. Viral particles were collected 48 hours posttransfection and concentrated by ultracentrifugation at 30 000g for 2 hours. Titering of final viral stock virus was performed at the Fred Hutchinson Cancer Research Center.36 HT1080 cells were plated in Dulbecco’s modified Eagle medium growth medium in 12-well plates. The following day, cells were transduced with dilutions of the viral vector. The cells were subsequently kept in culture for 10 days, during which they were passaged at a 1:5 split twice. DNA was extracted from cells, and the percentage of viral genome copies was analyzed by quantitative polymerase chain reaction.17
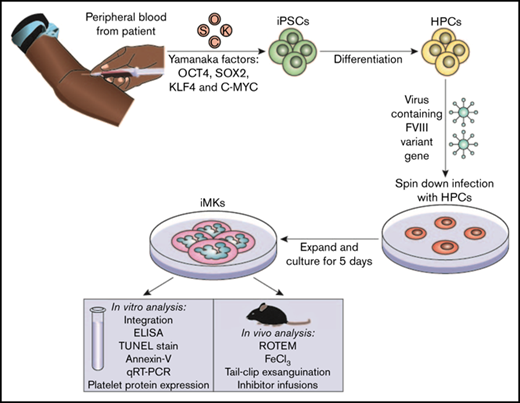
The iPSC line CHOPWT6 used in these studies for differentiation to iMKs has been previously described.37,38 The establishment and analysis of pFVIII-expressing iMKs are shown in the visual abstract. Hematopoietic progenitor cells (5 × 105 to 10 × 105 per well) were transfected with the FVIII-expressing lentiviral vectors at a multiplicity of infection of 1 to 10 via spin infection at 2000g for 2 hours at 37°C in a 12-well plate (BD Pharmingen) coated with 10 to 20 µg/cm−2 of RetroNectin (Takara Bio) in MK differentiation media.37,39 Viral particles were washed off the cells 24 hours postinfection. Cells were placed in fresh media for 5 days, and the resulting pFVIII-expressing iMKs were characterized in vitro and in vivo.
In vitro and in vivo studies of pFVIII-expressing iMKs
In vitro studies of pFVIII-expressing iMKs were performed by adding 10 µL of medium containing 5 × 103 to 5 × 105 pFVIII-expressing iMKs expressing hBDFVIII or the mutant hBDFVIIIRH, which has been shown by our group and others to have increased specific activity and hemostatic efficacy,17,40 to 110 µL of FVIIInull mouse blood. Negative controls were iMKs transfected with the eGFP-expressing lentivirus or PBS (Gibco) added to the same volume of blood. ROTEM studies were performed as described with line h38 whole blood with or without rFVIIa.
In vivo FeCl3 carotid artery injury studies using FVIIInull mice after xenotransfusion of human iMKs were similar to the studies with line h38 platelets, except that the recipient FVIIInull mice were pretreated with clodronate liposomes (Encapsula NanoSciences) 24 hours before iMK infusion to deplete macrophages.26 Similarly, tail clip at 1.6-mm diameter exsanguination studies were performed with iMKs infused in place of h38 platelet infusions as described.29 These were also performed in the presence of infused inhibitors. The percentage of human platelets circulating post-iMK infusion were determined after retroorbital blood draws by flow cytometry using species-specific anti-αIIb antibodies, as we have previously described.41 Recipient FVIIInull mice received 5 × 106 to 10 × 106 iMKs expressing pFVIII, pFVIIIRH, or eGFP via the jugular vein in 200-µL total volume of PBS. In some studies, rFVIIa was infused via the jugular vein at a concentration of 0.25 to 4.0 µg/kg concurrent with the iMKs.
Statistical analysis
Statistical differences between arms were determined using a 2-sided Student t test or analysis of variance with Bonferroni, Dunnett’s, or Tukey’s correction of multiple comparisons when appropriate. PRISM 7.0 (GraphPad) was used to calculate statistical significance. P ≤ .05 was considered significant.
Study approval
All animal procedures were approved by the Animal Care and Use Committee at the Children’s Hospital of Philadelphia in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. Human blood was collected after signed informed consent from healthy donors. Approval for studies using human blood was obtained from the Children’s Hospital of Philadelphia Institutional Human Review Board in accord with the Declaration of Helsinki principles.
Additional materials and methods
Supplemental Materials and methods provide details of the antibodies used (supplemental Table 1), viral integration analysis of iMKs, FVIII antigen–expression assays in iMKs, surface antigen studies on iMKs, iMK apoptosis studies, and responsiveness to thrombin.
Results
In vitro studies using line h38 platelets
We propose that infusions of pFVIII-containing platelets may be an effective alternative to BMT in patients with severe hemophilia A and intractable inhibitors for acute and/or prophylactic care. These platelets would provide coverage for several days and may provide additive therapeutic potential to a bypassing agent. As proof of principle for this concept and to simulate infusion of pFVIII platelet therapy, we used line h38 platelets which have ∼0.09 U (∼75 ng) of hBDFVIII antigen per milliliter of mouse blood.29 Previous in vitro studies were performed using ROTEM technology, which has shown that supplemental rFVIII can correct the hemostatic deficiency of FVIIInull blood.32,33,42 Whole blood from FVIIInull mice expressing high levels of pFVIII, equivalent to ∼0.2 U/mL of murine blood, has been reported to partially correct hemostatic function on thromboelastography, although prolonged clotting time was still shown.13,20 We confirmed these prior findings using line h38 whole blood. Compared with the positive control of wild-type (WT) murine blood and the negative control of FVIIInull murine blood, line h38 blood partially corrected the hemostatic defect of FVIIInull murine blood (Figure 1A). To determine the dose-dependency amount of line h38 blood that was required for this effect, line h38 whole blood was diluted with FVIIInull whole blood in different proportions, and ROTEM was performed (Figure 1B). Improved hemostasis was detectable with as little as 1% line h38 blood, and 20% line h38 blood was required to obtain results similar to those obtained with near-undiluted whole line h38 blood. By using rFVIII in this system, a dose of 0.01 ng/mL improved hemostatic function comparably to the level detected with 1% line h38 blood (supplemental Figure 2). Increasing doses of 0.1 ng/mL and 1.0 ng/mL of rFVIII increased hemostatic levels to those comparable to WT blood.
![Figure 1. ROTEM analysis of WT, line h38, and FVIIInull mice whole blood. (A) Whole blood from WT, line h38, and FVIIInull (knockout [KO]) mice were collected via the vena cava vein; ROTEM studies were then performed on those samples. Each curve represents 5 independent studies. (B) Same analysis as panel A; line h38 whole blood was diluted in FVIIInull to result in a 1%, 5%, or 20% level of h38 platelets in the blood mixture. Each curve represents 4 independent studies.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/9/10.1182_bloodadvances.2017007914/2/m_advances007914f1.png?Expires=1771146955&Signature=il00Q06RZ0CLQsknrNTtjxirlAkRddi5h9FjDa781rQFRNPo5ahsF3SFR715YIoC3ZeDwZAkfH-FlRFDjY88aFqU2RIjuQb0sCqI7NVKBWpahUcOKtjSMRxKP0~golHWjC0HczwzycVFdjB~~4XhjM4q7JtnC8jlneQ-FzirKupxlmnKLFPY23AiJTtAAP4VJ3oB-O4v78BLZFXIo4TcBCw04fGpVe8NAJyQKe~1LDXPZ3vOs3w22OexHuv6QKUZBZ75wUko1LkZEo~Tpe6Fe-gloif4nTLle5dmLKY9YKHaI4VHfGxZeCe1Sa3dAxVhnHsmPXdhU5LQQEOjgTd9oA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
ROTEM analysis of WT, line h38, and FVIIInullmice whole blood. (A) Whole blood from WT, line h38, and FVIIInull (knockout [KO]) mice were collected via the vena cava vein; ROTEM studies were then performed on those samples. Each curve represents 5 independent studies. (B) Same analysis as panel A; line h38 whole blood was diluted in FVIIInull to result in a 1%, 5%, or 20% level of h38 platelets in the blood mixture. Each curve represents 4 independent studies.
ROTEM analysis of WT, line h38, and FVIIInullmice whole blood. (A) Whole blood from WT, line h38, and FVIIInull (knockout [KO]) mice were collected via the vena cava vein; ROTEM studies were then performed on those samples. Each curve represents 5 independent studies. (B) Same analysis as panel A; line h38 whole blood was diluted in FVIIInull to result in a 1%, 5%, or 20% level of h38 platelets in the blood mixture. Each curve represents 4 independent studies.
We then compared the efficacy of pFVIII and rFVIIa. Improved hemostatic function was detectable with 10 nM of rFVIIa, whereas the addition of 25 nM of FVIIa was required to improve function comparable to WT levels, although with a delay (Figure 2A; supplemental Table 2). This delay is consistent with rFVIIa not directly correcting the underlying hemostatic defect, but rather bypassing the defective step in the coagulation cascade. The addition of 10 nM of rFVIIa to line h38 blood showed an additive effect with a decrease in clotting time; however, the lag in clotting time was still present (Figure 2B; supplemental Table 2). This failure to correct clotting time may be due to the time needed for platelet activation and pFVIII release.
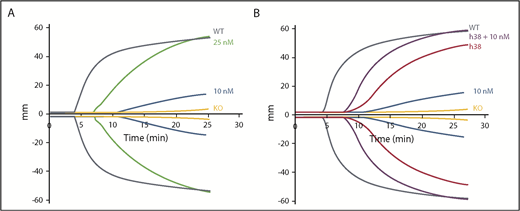
Additive effect of rFVIIa and pFVIII by ROTEM. ROTEM analyses were performed as in Figure 1. (A) rFVIIa was added to FVIIInull (KO) mouse whole blood at final concentrations of 10 and 25 nM. (B) Line h38 whole blood in the absence or presence of addition of 10 nM of rFVIIa was compared with FVIIInull mouse whole blood with addition of 10 nM of rFVIIa. WT and FVIIInull mouse blood were used as positive and negative controls, respectively. Each curve represents 4 independent studies. Supplemental Table 2 summarizes a parametric analysis of these data.
Additive effect of rFVIIa and pFVIII by ROTEM. ROTEM analyses were performed as in Figure 1. (A) rFVIIa was added to FVIIInull (KO) mouse whole blood at final concentrations of 10 and 25 nM. (B) Line h38 whole blood in the absence or presence of addition of 10 nM of rFVIIa was compared with FVIIInull mouse whole blood with addition of 10 nM of rFVIIa. WT and FVIIInull mouse blood were used as positive and negative controls, respectively. Each curve represents 4 independent studies. Supplemental Table 2 summarizes a parametric analysis of these data.
In vivo studies of infused line h38 platelets
We studied the hemostatic effects of infusing pFVIII-expressing line h38 platelets into FVIIInull recipient mice sufficient to result in ∼15% of the circulating platelets being from line h38. Such a level of infused platelets should be achievable clinically, and a correction of this level had shown hemostatic efficacy in vitro (Figure 1). These infused mice survived an overnight tail-clip exsanguination assay, which, in agreement with prior observations,14 was 100% lethal in nontransfused FVIIInull mice or mice infused with non-pFVIII platelets. Beginning at 1 and extending to 72 hours postinfusion of line h38 platelets (Figure 3A; supplemental Table 3), there was improved survival in this tail-clipping hemostatic model in FVIIInull mice. This effect was not due to simply having infused platelets into the FVIIInull mice (Figure 3A). We also found that the half-life of infused platelets prepared by us was ∼22 hours (supplemental Figure 3), consistent with half-lives reported for infused murine platelets by others.41,43,44 By 72 hours postinfusion of 15% line h38 platelets, we anticipated that <2% of the circulating platelets would be line h38 derived. In contrast to this infusion of pFVIII platelets, infusion of rFVIIa was ineffective at rescuing recipient FVIIInull mice from tail-clip exsanguination even 0.25 hour postinfusion, unless a high dose of 4 mg/kg of rFVIIa was administered; however, even then, hemostatic efficacy was mostly gone by 1.5 hours postinfusion (Figure 3B; supplemental Table 3), consistent with the short half-life of human rFVIIa in mice.11
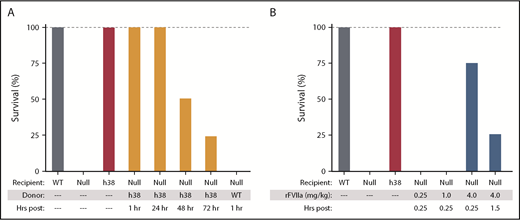
Survival after line h38 platelets or pFVIII-expressing iMKs and rFVIIa infusion in the tail-clip exsanguination assay. (A-B) Tail-clip exsanguination assay with expiration assessed 16 hours post–tail clip. In both panels, on the left, WT, FVIIInull, and line h38 mice underwent tail-clip exsanguination. Percentage of animals surviving of 5 animals per arm is shown. Dashed gray line is 100% survival. (A) On the right, FVIIInull mice were infused to ∼15% line h38 platelets and then underwent tail-clip exsanguination at 1 to 72 hours postinfusion. (B) On the right, mice were infused with 0.25, 1, and 4 mg/kg of rFVIIa retroorbitally 15 minutes before tail resection; additional mice were studied at 4 mg/kg of rFVIIa 1.5 hours postinfusion.
Survival after line h38 platelets or pFVIII-expressing iMKs and rFVIIa infusion in the tail-clip exsanguination assay. (A-B) Tail-clip exsanguination assay with expiration assessed 16 hours post–tail clip. In both panels, on the left, WT, FVIIInull, and line h38 mice underwent tail-clip exsanguination. Percentage of animals surviving of 5 animals per arm is shown. Dashed gray line is 100% survival. (A) On the right, FVIIInull mice were infused to ∼15% line h38 platelets and then underwent tail-clip exsanguination at 1 to 72 hours postinfusion. (B) On the right, mice were infused with 0.25, 1, and 4 mg/kg of rFVIIa retroorbitally 15 minutes before tail resection; additional mice were studied at 4 mg/kg of rFVIIa 1.5 hours postinfusion.
The FeCl3 carotid artery injury model was also used to study in vivo hemostatic challenges in FVIIInull mice. Previous work from our group has shown a strong positive correlative relationship between infused human rFVIII into FVIIInull mice and improved hemostasis.29 This hemostatic challenge can be completed within 30 minutes and thus would allow studies of the additive effects of pFVIII with rFVIIa. Infusion of line h38 platelets, but not non-pFVIII platelets, into FVIIInull mice was effective in decreasing blood flow through the injured vessel up to 48 hours postinfusion, as measured by area under the curve (Figure 4A). rFVIIa was also effective in improving hemostasis in this model, and an additive effect with line h38 platelets was seen in the recipient FVIIInull mice (Figure 4B).
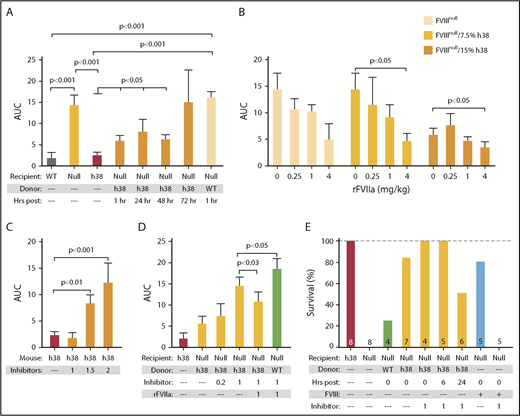
Efficacy of line h38 platelet hemostatic efficacy in mice. (A-D) FeCl3 carotid artery injury studies were as described.17 Area under the curve (AUC) of subsequent blood flow was measured. In all studies, P values were determined by 1-way analysis of variance. (A) WT, FVIIInull, and line h38 mice were studied, as were FVIIInull mice infused with either WT or line h38 platelets to achieve ∼20% of the circulating platelets in the recipient. Animals infused with line h38 platelets were studied up to 72 hours later. Mean ± 1 standard error of the mean (SEM) are shown, with 5 animals per arm. (B) Same as in panel A, with studied FVIIInull mice infused with the indicated amounts of line h38 platelets and rFVIIa. Mean ± 1 SEM are shown, with 5 animals per arm. (C) Line h38 mice were infused with varying concentrations of the inhibitor mixture (in milligrams per kilogram of mouse) before the FeCl3 injury. Mean ± 1 SEM are shown, with 6 animals per arm. (D) FVIIInull and line h38 mice were studied in the FeCl3 carotid artery injury model, as in panel A. FVIIInull mice were also infused with ∼15% line h38 or WT platelets and with varying concentrations of the inhibitor mixture (in milligrams per kilogram of mouse) ± 1 mg/kg of rFVIIa before the FeCl3 injury. Mean ± 1 SEM are shown, with 5 animals per arm. (E) Tail-clip exsanguination studies, as in Figure 3. Mice received either h38 platelets or human FVIII infusions at 0.125 U/mL of blood. Tail-clip exsanguination studies were delayed for 0 to 24 hours after h38 platelet infusion as indicated. Number of mice studied is indicated in each bar.
Efficacy of line h38 platelet hemostatic efficacy in mice. (A-D) FeCl3 carotid artery injury studies were as described.17 Area under the curve (AUC) of subsequent blood flow was measured. In all studies, P values were determined by 1-way analysis of variance. (A) WT, FVIIInull, and line h38 mice were studied, as were FVIIInull mice infused with either WT or line h38 platelets to achieve ∼20% of the circulating platelets in the recipient. Animals infused with line h38 platelets were studied up to 72 hours later. Mean ± 1 standard error of the mean (SEM) are shown, with 5 animals per arm. (B) Same as in panel A, with studied FVIIInull mice infused with the indicated amounts of line h38 platelets and rFVIIa. Mean ± 1 SEM are shown, with 5 animals per arm. (C) Line h38 mice were infused with varying concentrations of the inhibitor mixture (in milligrams per kilogram of mouse) before the FeCl3 injury. Mean ± 1 SEM are shown, with 6 animals per arm. (D) FVIIInull and line h38 mice were studied in the FeCl3 carotid artery injury model, as in panel A. FVIIInull mice were also infused with ∼15% line h38 or WT platelets and with varying concentrations of the inhibitor mixture (in milligrams per kilogram of mouse) ± 1 mg/kg of rFVIIa before the FeCl3 injury. Mean ± 1 SEM are shown, with 5 animals per arm. (E) Tail-clip exsanguination studies, as in Figure 3. Mice received either h38 platelets or human FVIII infusions at 0.125 U/mL of blood. Tail-clip exsanguination studies were delayed for 0 to 24 hours after h38 platelet infusion as indicated. Number of mice studied is indicated in each bar.
We further tested the efficacy of prophylactic pFVIII infusions in the presence of inhibitors. Before platelet infusion, we injected mice with a single dose of a 1:5 inhibitor mixture in micrograms of ESH8 (known to bind to the FVIII C domain45 ) and GMA-8021 (known to bind to the FVIII A2 domain),46 which we have previously shown to effectively inhibit, in combination, pFVIII function in line h38 mice in the FeCl3 carotid artery thrombosis model.15 We confirmed these findings and determined that an inhibition dose of 1 mg of total inhibitor cocktail per kilogram of mouse did not inhibit hemostasis in line h38 mice (Figure 4C). This same inhibitor dose was able to partially inhibit the effectiveness of an infusion of line h38 platelets to 15% in FVIIInull mice (Figure 4D) relative to infused WT platelets that did not express pFVIII. The addition of rFVIIa to this pFVIII therapy in the presence of an inhibitor was able to further enhance hemostasis.
We also examined whether the infused line h38 platelets would be protective in the presence of the inhibitor mixture (1 mg per kilogram of mouse) using the more sensitive tail-clip exsanguination assay, as in Figure 3, and found that the pFVIII-containing platelets were protective even if administered 24 hours before the tail clip and even in the presence of the inhibitor, whereas infusion of FVIII to achieve a comparable whole blood level was not protective when the inhibitor was also infused (Figure 4E).
Establishing and characterizing human pFVIII-expressing iMKs
We then wanted to expand these studies using human platelets expressing pFVIII. We used an iPSC line that we previously reported could be differentiated into a significant number of evaluable iMKs.37,39 The iPSC-derived hematopoietic progenitor cells were transduced with the previously described Cxcl4-proximal promoter-driven hBDFVIII lentiviral vectors, hBDFVIII or hBDFVIIIRH, or eGFP.16,17 A schematic showing iPSC hematopoietic differentiation, FVIII lentiviral transduction, and downstream applications is shown in the visual abstract. On day 6 of differentiation, lentiviral copy numbers of transduced iMKs were studied, and no significant difference between the 3 vectors was shown, although the highest level was seen for the hBDFVIIIRH iMKs (supplemental Figure 4A). F8 mRNA was highest in hBDFVIII iMKs (P < .001; supplemental Figure 4B); in contrast, FVIII antigen per cell by enzyme-linked immunosorbent assay was ∼50% higher in hBDFVIIIRH iMKs (P < .01; supplemental Figure 4C).
Our previous studies have suggested that ectopically expressed FVIII in MKs was harmful to the differentiating cells.17 To determine if damage to iMKs occurred in this system, examination of cells for annexin V binding (supplemental Figure 5A), bromodeoxyuridine staining via terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling assay (supplemental Figure 5B), and examination of percentage of the CD41+ pFVIII-expressing iMKs that were also CD42b+ (an indicator of undamaged MKs; supplemental Figure 5C) were performed, comparing pFVIII-expressing iMKs with nontransfected iMK controls and showing no difference in health. In addition, the health of the pFVIII-expressing iMKs was analyzed by responsiveness to agonist stimulation, and no difference was observed (supplemental Figure 5D). The health of these iMKs was likely due to human pFVIII not being as injurious to MKs as canine FVIII, and the level of human pFVIII expressed in these iMKs was low. High levels of canine FVIII expressed in MKs are toxic to expressing cells,17,47 and the levels of human pFVIII achieved in our study were only ∼1% of those seen with line h38 transgenic mice MKs29 (supplemental Figure 4C).
Hemostatic studies with human pFVIII-expressing iMKs
ROTEM studies were performed with pFVIII-expressing iMKs added to FVIIInull murine whole blood and demonstrated that as few as 5 × 103 pFVIII-expressing iMKs added to 110 µL of whole blood could improve hemostasis compared with the addition of iMKs not expressing FVIII (Figure 5A-B). The hBDFVIIIRH iMKs showed greater efficacy than hBDFVIII iMKs, as indicated by shorter clotting time and increased α angle (Figure 5A-B; supplemental Table 4). The addition of 10 nM of rFVIIa showed a combinatorial effect with the pFVIII-expressing iMKs, further improving hemostasis although not correcting the clotting time (Figure 5C; supplemental Table 5), supporting the line h38 platelet data shown in Figure 1.
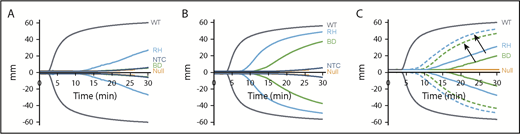
In vitro efficacy of pFVIII-expressing iMKs in ROTEM studies. ROTEM studies, as in Figure 1, but with added hBDFVIII (BD), hBDFVIIIRH (RH), or nontransfected control (NTC) iMKs instead of line h38 platelets. Each curve represents 5 independent studies. (A) Addition of 5 × 103 iMKs to 110 µL of whole blood from FVIIInull (KO) mice. WT and FVIIInull mice blood were studied as positive and negative controls, respectively. (B) Same as in panel A, with 5 × 104 BD or RH iMKs. (C) Same as in panel A, but with (dotted lines) or without (solid lines) 10 nM of rFVIIa added. Arrows indicate paired studies with or without rFVIIa. Supplemental Table 4 and supplemental Table 5 summarize parametric analyses of these data for Figure 6A-B and Figure 6C, respectively.
In vitro efficacy of pFVIII-expressing iMKs in ROTEM studies. ROTEM studies, as in Figure 1, but with added hBDFVIII (BD), hBDFVIIIRH (RH), or nontransfected control (NTC) iMKs instead of line h38 platelets. Each curve represents 5 independent studies. (A) Addition of 5 × 103 iMKs to 110 µL of whole blood from FVIIInull (KO) mice. WT and FVIIInull mice blood were studied as positive and negative controls, respectively. (B) Same as in panel A, with 5 × 104 BD or RH iMKs. (C) Same as in panel A, but with (dotted lines) or without (solid lines) 10 nM of rFVIIa added. Arrows indicate paired studies with or without rFVIIa. Supplemental Table 4 and supplemental Table 5 summarize parametric analyses of these data for Figure 6A-B and Figure 6C, respectively.
In vitro–grown human MKs from various sources have been used to generate platelets both by direct harvesting of platelet-like particles (PLPs) in static culture48-50 or by using various bioreactors.25,51-53 Many of these PLPs lack surface CD41 and are not derived from MKs. Only a small percentage of CD41+ PLPs are noninjured annexin V−/CD42b+ platelets. Not surprisingly, these PLPs have limited half-life in circulation postinfusion, are preactivated, or have poor agonist responsiveness. In contrast, platelets released from IV infusion of human MKs, in which the MKs shed platelets within the lungs over the subsequent few hours,26 are more physiologically intact, similar to platelets released from endogenous murine MKs entrapped in the lungs.54 The released human platelets have a Gaussian-size distribution, circulating half-life, and agonist responsiveness similar to well-prepared, fresh, donor-derived platelets.26,41 At present, there is no available FVIIInull murine model on an immune-compromised background available for human MK infusion studies, so we infused iMKs into clodronate liposome–treated FVIIInull mice to improve human platelet half-life by reducing phagocytic removal of the human platelets.26,55 Infused iMKs released detectable human platelets with a half-life of ∼4 hours (Figure 6A). In FeCl3 carotid artery studies initiated within 30 minutes after iMK infusion, both hBDFVIII and hBDFVIIIRH iMKs improved hemostasis significantly compared with non–pFVIII-expressing iMKs, although none of these mice developed occlusive thrombi (Figure 6B). As in the ROTEM studies, hBDFVIIIRH iMKs improved hemostasis better than hBDFVIII iMKs. We carried out iMK infusion studies similar to those in Figure 6B and examined survival in the tail-clip exsanguination assay, which is more sensitive to the hemostatic effect of pFVIII, similar to Figure 4E but with coinfused inhibitor. hBDFVIIIRH iMKs, but not the nontransfected control iMKs, could rescue the mice from overnight tail-clip exsanguination both in the absence and presence of the inhibitor (Figure 6C).
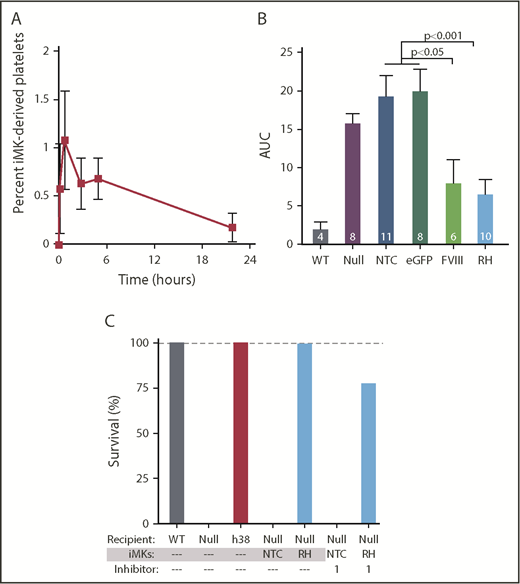
pFVIII-expressing iMK infusion hemostatic efficacy in FVIIInullmice. (A-C) 5 × 106 to 10 × 106 iMKs were infused into clodronate liposome–treated FVIIInull mice. (A) Human platelets released from infused iMKs were analyzed up to 24 hours after iMK infusion via flow cytometry. Mean ± 1 SEM are shown, with 5 independent studies. (B) Similar FeCl3 carotid artery injury studies as in Figure 4, with the primary end point being remaining blood flow as measured by AUC. Mean ± 1 SEM are shown, with the numbers of independent studies noted in the bars. P values were determined by 1-way analysis of variance. (C) Tail-clip exsanguination studies as in Figure 3, but for pFVIII-expressing iMK infusions. The 3 left-most bars represent 5 mice per group. The 4 right-most bars represent nontransfected control (NTC) or hBDFVIIIRH (RH) iMKs infused into FVIIInull mice, with 5 animals per arm in the presence of 1 mg/kg of inhibitor per mouse. Dashed gray line is 100% survival.
pFVIII-expressing iMK infusion hemostatic efficacy in FVIIInullmice. (A-C) 5 × 106 to 10 × 106 iMKs were infused into clodronate liposome–treated FVIIInull mice. (A) Human platelets released from infused iMKs were analyzed up to 24 hours after iMK infusion via flow cytometry. Mean ± 1 SEM are shown, with 5 independent studies. (B) Similar FeCl3 carotid artery injury studies as in Figure 4, with the primary end point being remaining blood flow as measured by AUC. Mean ± 1 SEM are shown, with the numbers of independent studies noted in the bars. P values were determined by 1-way analysis of variance. (C) Tail-clip exsanguination studies as in Figure 3, but for pFVIII-expressing iMK infusions. The 3 left-most bars represent 5 mice per group. The 4 right-most bars represent nontransfected control (NTC) or hBDFVIIIRH (RH) iMKs infused into FVIIInull mice, with 5 animals per arm in the presence of 1 mg/kg of inhibitor per mouse. Dashed gray line is 100% survival.
Discussion
We have tested whether infusion of platelets that can deliver pFVIII could be a potential strategy for treating patients with severe hemophilia A and intractable inhibitors. We provide proof of principle both in in vitro and in vivo hemostatic models that infusion of pFVIII-expressing murine platelets can improve hemostasis. For infused pFVIII murine platelets, the hemostatic effect lasted several days, even though the murine platelet half-life is half that of human platelets.41,56 This hemostatic effect was seen with inhibitors present and additive with the hemostatic effects of rFVIIa, suggesting that the 2 therapies can be combined for higher efficacy. Studies with pFVIII-expressing iMks support these results and suggest that once robust technologies are developed to generate clinically relevant human platelets, whether from iPSCs or from another cell line, such as adipose cells,57 they could have wider application beyond just providing an alternative source of platelet transfusions; they could provide a delivery system for various therapeutic agents, perhaps beginning with pFVIII in the described clinical settings.
One scenario for pFVIII infusion therapy could be a patient with intractable inhibitors with breakthrough bleeding receiving emicizumab to avoid the thrombotic risks of bypass agents. Such a scenario would supplement the hemlibra with universal pFVIII-expressing iMKs, which would release platelets missing particular HLA-related antigens on their surface.58 We envision the patient requiring only weekly infusions, assuming the transfused platelets lasted at least 1 week, with limited increases in total platelet counts. Applying this strategy to pFVIII delivery would provide insights into what type of clinically relevant bleeds pFVIII would be useful for and whether in a particular patient with specific inhibitors there would be benefit from a more permanent pFVIII therapy using marrow-directed gene therapy. Furthermore, the pFVIII platelets may then be useful to support such patients during their post-BMT thrombocytopenia, with its increased bleeding risks, perhaps along with emicizumab, avoiding FVIII bypass agents that might enhance prothrombotic risks.
The presented studies with line h38 pFVIII platelets have several limitations. One major limitation is the low levels of pFVIII being studied. Line h38 transgenic mice have the whole-blood equivalency of 3% FVIII plasma activity (human FVIII has ∼30% activity within murine plasma16,18,29 ), with all of the FVIII sequestered within platelets.16,29 In the presented in vivo studies, where line h38 platelets represented ≤15% of the total platelets, the blood would have ≤0.6% equivalency of FVIII activity. This low level of FVIII may have contributed to the suboptimal contraction seen in the ROTEM studies (Figure 1), in addition to the need for platelet activation to release pFVIII relative to the instant availability of plasma FVIII. In the tail-clip exsanguination assay, the efficacy of pFVIII at such low levels is consistent with our prior proposal that the tail-clip exsanguination assay is especially sensitive to pFVIII.59 Calculated levels of plasma-equivalent FVIII activity as low as 0.02% were effective 3 days after line h38 platelet infusion in this hemostatic challenge (Figure 3). Finally, the low pFVIII levels may explain why such a high concentration of sclerosing FeCl3 was needed in the carotid artery studies, with the end point of decreased arterial blood flow rather than full occlusion (Figure 4). Thus, if only low levels of pFVIII can be achieved in patients, its clinical value may be limited, especially in the presence of high-titer inhibitors. Whether that low level would still be beneficial and useful in conjunction with rFVIIa would also need to be addressed clinically.
The pFVIII-expressing iMK studies also have several important limitations. iMKs are known to be small, with low ploidy, and release ∼10 platelets per iMK, even upon infusion into recipient mice.26 Moreover, the survival of the released human platelets in clodronate liposome–pretreated FVIIInull mice (Figure 6A) was shorter than the ∼12 hours seen when human iMKs were infused into immunodeficient mice.26 In addition, the level of pFVIII in these iMKs was even lower than that of the line h38 platelets; clearly a strategy to achieve higher levels of pFVIII per iMK needs to be established. We have described such a strategy using adeno-associated virus site 1 targeting of vector constructs to drive MK-specific expression using a Gp1ba promoter, but although this strategy achieved high levels of eGFP expression and correction of αIIb expression in iMKs,39 it did not drive significant expression of pFVIII (data not shown).
Remarkably, even with the low level of pFVIII in the studied iMKs, the low number of platelets released per iMK, and the shortened released human platelet half-life, efficacy in the FeCl3 carotid artery injury model was seen with hBDFVIIIRH iMKs (Figure 6B). Better hemostasis with hBDFVIIIRH iMKs compared with hBDFVIII iMKs was also seen in the ROTEM studies (Figure 6). Although hBDFVIIIRH pFVIII levels were slightly higher than hBDFVIII pFVIII levels in iMKs, this difference was small (supplemental Figure 3C) and not likely responsible for the greater efficacy of hBDFVIIIRH pFVIII. Moreover, this finding is also consistent with our previous studies showing that in FVIIInull mice expressing hBDFVIIIRH FVIII after lentiviral gene therapy, there was greater efficacy in improving hemostasis in the cremaster arteriole laser injury model because of increased hBDFVIIIRH stability within the core of the growing thrombus.16,17,59
In summary, we demonstrate an alternative to BMT, using a pFVIII-based strategy to treat patients with severe hemophilia A and intractable inhibitors. This approach may provide a means to avoid bypassing strategies and provide effective FVIII in these patients. It has the advantage of not permanently altering the patients and can potentially provide longer-lasting prophylaxis and be used in conjunction with other bypass therapies. It may also provide insights into the benefits of platelet FVIII storage before attempting permanent pFVIII gene therapy to demonstrate efficacy in the presence of a patient’s particular inhibitors and specific target organs. It may also be a useful adjuvant for bleeding challenges after marrow transplantation for gene therapy inpatients with inhibitors. Finally, it may be a first-to-clinic application of in vitro–generated MKs and derived platelets that would justify the costs of preparing such platelets relative to donor-derived platelets.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Martin E. Wohlfahrt and Beverly Torok-Storb at the Fred Hutchinson Cancer Research Center for titering of our viral stocks.
This work was supported by grants T32HL007971 (R.B.L.), U01HL099656 (M.P.), and R01HL132557 (M.P.) from the National Heart, Lung, and Blood Institute, National Institutes of Health, and by a Pfizer ASPIRE Hemophilia Research Award (S.K.S.).
Authorship
Contribution: R.B.L. carried out and evaluated the studies and prepared the first draft of the manuscript; H.S.A., K.K.V., J.T., and D.J.J. helped carry out and complete experiments; E.T. prepared illustrations; S.K.S., D.E.S., D.L.F., and R.M.C. provided equipment for the studies and edited and made intellectual contributions to the manuscript; and M.P. provided overall project organization and direction, data interpretation, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mortimer Poncz, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, ARC, Room 317, Philadelphia, PA 19104; e-mail: poncz@email.chop.edu.