

Abstract
Clinical outcomes for children with acute myeloid leukemia (AML) have improved minimally during the past 4 decades despite maximally intensive chemotherapy, hematopoietic stem cell transplantation, and optimized supportive care. Chemoresistance and relapse remain major sources of childhood cancer–associated mortality and highlight the need for alternative treatment approaches. The remarkable recent success of humoral and cellular immunotherapies in children and adults with relapsed/refractory B-acute lymphoblastic leukemia has inspired hope for similar accomplishments in patients with AML. However, unique challenges exist, including the biologic and immunophenotypic heterogeneity of childhood AML and the significant potential for on-target/off-tumor immunotherapeutic toxicity due to target antigen expression on nonmalignant cells. This article reviews the current landscape of antibody-based and cellular immunotherapies under current clinical evaluation with an emphasis on active or soon-to-open phase 1 trials for children with relapsed/refractory AML.
Introduction
Development of immunotherapies for children and adults with acute myeloid leukemia (AML) has been fraught with challenges, including lack of identified tumor-specific antigens, inter- and intrapatient disease heterogeneity, and increased recognition of immunosuppressive bone marrow microenvironment factors that have hindered therapeutic success.1-3 In theory, an ideal antigen for immunotherapeutic targeting is universally and highly expressed on tumor cells, particularly cancer-initiating cells, but is absent in normal tissues. In practice, such antigens are rarely discovered, and immunotherapeutic strategies thus aim to maximize a “therapeutic window” of robust antitumor activity with minimal effects on antigen-bearing nonmalignant cells. Although CD19 indeed appears to be a “universal” tumor antigen in patients with B-cell acute lymphoblastic leukemia (B-ALL) and aplasia of normal B cells, a clinically tolerable on-target/off-tumor sequela manageable with immunoglobulin infusion supportive care, most antigens of potential immunotherapeutic interest in AML are also expressed on hematopoietic stem and/or myeloid progenitor cells. Targeting of such antigens theoretically risks prolonged or permanent marrow aplasia bystander toxicity that may require subsequent hematopoietic stem cell transplantation (HSCT) rescue.
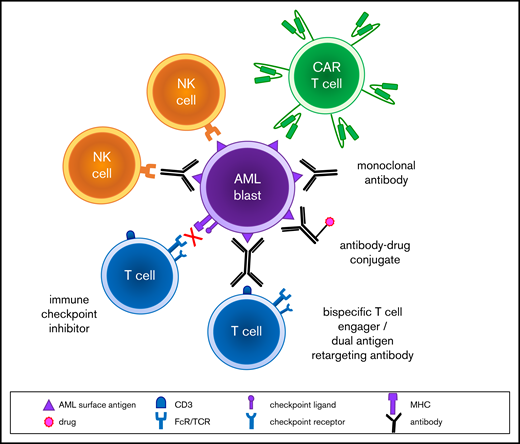
Modern molecular diagnostic testing via next-generation sequencing platforms has significantly improved understanding regarding risk stratification and prognosis of children with AML.2 These data have facilitated precision medicine treatment approaches for small subsets of patients for whom targeted inhibitors are available, such as sorafenib addition to chemotherapy for children with newly diagnosed FLT3 (FMS-like tyrosine kinase 3)-mutant AML (Children’s Oncology Group [COG] trial AAML1031; NCT01371981) or trametinib therapy for children with relapsed RAS pathway–mutant juvenile myelomonocytic leukemia (COG ADVL1521; NCT03190915). Several genetic subtypes of childhood AML are now known to be associated with unique flow cytometric immunophenotypes,4 which may provide further opportunities to individualize therapy. Given the biologic and genetic heterogeneity of childhood AML, it is likely that multiple immunotherapies targeting a variety of tumor antigens must be successfully developed to improve cure rates appreciably (Figure 1). We describe 3 patient case scenarios below with a goal of illustrating how immunotherapeutic strategies can be incorporated into the care of children with high-risk AML.
Clinical case 1
A 7-year-old boy was diagnosed with AML after presenting with progressive fatigue, easy bruising, and splenomegaly. Cytogenetic and fluorescence in situ hybridization analysis of his bone marrow demonstrated KMT2A-MLLT3 fusion from t(9;11). The child was induced with cytarabine, daunorubicin, and etoposide (ADE) as per the COG AAML0531 (NCT00372593) and AAML1031 (NCT01371981) phase 3 studies, and he had no evidence of minimal residual disease (MRD) by flow cytometry after the first induction cycle. He received a total of 5 cycles of multiagent chemotherapy and remained in clinical remission until 16 months off therapy, when routine complete blood count surveillance demonstrated thrombocytopenia and leukocytosis with peripheral blasts. Flow cytometric immunophenotyping of his relapse specimen showed bright CD33 surface expression concordant with a CD33 CC single-nucleotide polymorphism genotype. The child was reinduced with fludarabine and cytarabine with filgrastim support (FLAG)5 and one dose of gemtuzumab ozogamicin (GO), and a second MRD-negative remission (CR2) was achieved. He received an additional cycle of FLAG and underwent allogeneic HSCT from an HLA-matched sibling and did not have sinusoidal obstruction syndrome/veno-occlusive disease (SOS/VOD). He remains in continued MRD-negative remission with complete donor chimerism.
Role of HSCT for children with relapsed AML
Although most children with AML achieve initial remission induction with multiagent chemotherapy, relapse due to presumed chemoresistance remains a major source of childhood cancer–associated mortality and can be challenging to overcome with intensive salvage chemotherapy.6,7 In addition, a small percentage of children with AML (potentially arising from antecedent myelodysplastic syndromes, which are far less common in the pediatric vs adult population) have primary chemoresistance and are unable to achieve initial remission (CR1). Nearly all children who experience AML relapse and achieve CR2 after reinduction chemotherapy undergo subsequent allogeneic HSCT, which provides an opportunity for additional myeloablative chemotherapy administration to eradicate leukemia and to enhance potential for desirable graft-versus-leukemia (GVL) immune effects from donor T cells. This approach has achieved 5-year overall survival (OS) rates of approximately 40%, although outcomes are significantly worse for children who relapse within 1 year from initial AML diagnosis.8,9 Close posttransplantation monitoring of donor chimerism and for leukemia relapse is imperative because rapid withdrawal of immunosuppressive medications and/or donor lymphocyte infusion (DLI) has been reported to augment GVL effects in some patients with falling chimerism and suspicion for early AML recurrence.10 Potential benefit of DLI must be balanced carefully with risk of graft-versus-host disease (GVHD) from deleterious donor T-cell effects on normal recipient tissues, however. Unfortunately, many patients with low-level AML recurrence treated with DLI eventually experience overt relapse. Second transplantation, often from a different allogeneic donor and using an alternative conditioning regimen, is now being used for some patients who relapse after HSCT. The long-term success of such approaches remains limited and is influenced in part by proximity of relapse timing after transplantation.11-13
Several genetic subtypes of childhood AML are well known to be particularly unfavorable (eg, FLT3 internal tandem duplication, monosomy 7, monosomy 5/deletion 5q) and are associated with extremely poor event-free survival (EFS) with chemotherapy alone. Most cooperative groups have allocated such patients after 3 chemotherapy cycles to best-available donor allogeneic HSCT in CR1 with a goal of preventing relapse and maximizing GVL immune effects.6,7 Other groups have more sparingly used HSCT in this population, given its appreciable toxicity risk, particularly in the absence of clear data demonstrating improved OS with HSCT in CR1.14 A soon-to-open COG phase 3 trial (AAML1831) will assess a refined AML risk stratification based on genetic sequencing data and associated outcome data of clinical trials conducted by several consortia. It is estimated that up to half of children with de novo AML will be now be classified as high risk on the basis of leukemia-associated genetic alterations (Table 1) and/or positive end-induction MRD and will be allocated to HSCT in CR1.15 Larger studies are needed to ascertain whether this approach improves EFS and OS in all children with high-risk AML, particularly for those recently identified rare subtypes for which the potential benefit (or lack of benefit) of HSCT is unknown.2,7
GO: a Food and Drug Administration–approved immunotherapy for children with relapsed AML
CD33 is a transmembrane receptor in the sialic acid–binding immunoglobulin-like lectin family of proteins. It is expressed on the cell surface of >90% of childhood AML cases in varying intensity. High CD33 surface expression was associated with adverse disease characteristics and inferior clinical outcomes in children with newly diagnosed AML treated in the COG AAML0531 phase 3 clinical trial and, more recently, with greater incidence of the CD33 nonsynonymous coding single-nucleotide polymorphism rs12459419 CC wild-type genotype and favorable clinical responses to the CD33-targeting antibody–drug conjugate (ADC) GO.16,17 Conversely, children with AML and the TT genotype have truncated CD33 protein expression and lack the binding site for GO; these patients were noted to have inferior EFS in the AALL0531 trial.16 CD33 is also expressed on normal myeloid precursor cells and on hepatic Kupffer cells, which are important considerations for potential “on-target/off-tumor” sequelae of GO and other CD33-targeted immunotherapies, including SOS/VOS.18
GO is a CD33 ADC with a calicheamicin payload that induces cell death via DNA binding. GO received accelerated Food and Drug Administration (FDA) approval in 2000 but was withdrawn voluntarily from the market in 2010 because of toxicity and efficacy concerns. Early studies of GO reported an increased incidence in SOS/VOD likely due to higher GO dosing than is currently used and administration in close proximity to HSCT. Subsequent larger studies observed definitive clinical benefit and failed to demonstrate increased risk of SOS/VOD.17,19 Improved outcomes with GO addition to chemotherapy for children with AML were specifically observed in the AAML0531 trial with higher relapse-free survival in certain patient subsets.17 GO was reapproved in 2016 by the FDA and in 2018 by the European Medicines Agency for adults with relapsed or de novo AML. GO also was approved by the FDA for children 2 years of age and older with relapsed (but not de novo) AML. On the basis of AAML0531 results and other favorable outcome data, GO will be added to chemotherapy for all patients with newly diagnosed CD33+ AML in the forthcoming COG phase 3 trial.
Clinical case 2
A 2-year-old boy was diagnosed with acute megakaryoblastic leukemia after presenting with pallor and epistaxis. Bone marrow aspiration and biopsy demonstrated a fibrotic marrow with 30% patchy leukemia involvement. Flow cytometric immunophenotyping showed intermediate CD33, bright CD56, and absent HLA-DR surface expression concordant with the RAM phenotype.20 Cytogenetic and molecular analysis demonstrated inv(16) with CBFA2T3-GLIS2 fusion. The child received induction therapy with ADE and GO and had 12.5% MRD after cycle 1. Therapy was intensified to cytarabine and mitoxantrone as per COG AAML1031 (recently reported not to improve 5-year disease-free survival vs a second cycle of ADE21 ), and MRD was 15% after cycle 2. Given his demonstrated chemotherapy resistance and high-risk leukemia-associated genetic alteration, the boy was enrolled in a phase 1 clinical trial of autologous CD33-redirected chimeric antigen receptor (CAR) T-cell immunotherapy. He successfully underwent T-cell pheresis and manufacturing and was in morphologic remission with flow cytometric MRD <0.1% at 30 days after CD33 CAR T-cell infusion with antecedent lymphodepletion. The boy subsequently received an allogeneic HSCT from an HLA-matched unrelated donor and remains under close clinical surveillance for engraftment and relapse monitoring.
Cellular immunotherapies for children with relapsed/refractory AML
CAR T cells redirected against B-cell antigens (eg, CD19, CD22) have demonstrated remarkable clinical activity in children and adults with relapsed/refractory B-cell malignancies, leading to recent FDA approval of 2 CD19 CAR T-cell (CD19CART) immunotherapies. These products are usually manufactured from autologous T cells obtained from patients via apheresis. These T cells are lentivirally transduced ex vivo with CAR constructs comprised of high-affinity single-chain variable fragments typically derived from antibodies directed against target tumor antigens, transmembrane protein hinges, and intracellular costimulatory domains that confer robust T-cell expansion and persistence properties. Successful development of CAR T cells for patients with AML has been far more challenging, primarily owing to lack of identified “universal” myeloid antigens for therapeutic targeting and appreciable risk of “on-target/off-tumor” hematologic and nonhematologic toxicity.
On the basis of improved efficacy and clinical tolerability of GO and chemotherapy in children with AML, CD33 expression in the majority of pediatric AML cases, and promising preclinical data (reviewed in detail elsewhere22 ), several phase 1 studies have focused on clinical evaluation of CD33-targeted CAR T cells (CD33CART) in adults with relapsed/refractory AML (Table 2). Preliminary results of these trials have been largely restricted to single-patient case reports reported by investigators in China, who have noted partial or complete responses in some patients with a few successfully bridged to allogeneic HSCT. Importantly, these studies have demonstrated clinical tolerability with manageable cytokine release syndrome (CRS) and minimal SOS/VOD and myelosuppression.23,24 Additional US-based immunotherapy clinical trials are currently recruiting or in development that will allow participation of children and adolescents with AML. A phase 1 CD33CART trial at the MD Anderson Cancer Center is currently open for patients with second or greater relapsed/refractory AML (NCT03126864). This study is assessing safety and tolerability and will determine the recommended phase 2 dose of CD33CART in separate adult and pediatric strata (12-18 years of age). A planned phase 1 trial of interleukin (IL)-12–stimulated CD33CART at the Memorial Sloan Kettering Cancer Center will also treat a pediatric AML cohort. Finally, a first-in-child Pediatric Blood and Marrow Transplant Consortium–sponsored multi-institutional lentivirus/second-generation CD33CART phase 1 trial will soon open at the National Cancer Institute and the Children’s Hospital of Philadelphia for children and adolescents/young adults (1-30 years of age) with relapsed/refractory CD33+ AML.25
CD123 (IL-3 receptor α-chain) is another surface antigen expressed in the majority of childhood AML and, importantly, is reportedly also expressed on leukemia-initiating cells.26 CD123 is expressed on normal myeloid precursors and on cardiac endothelial cells at lower levels, suggesting theoretical potential for myelosuppression and cardiac toxicity of CD123 immunotherapy. CD123 is also highly expressed on B-ALL cells, which broadens potential clinical indications for CD123-targeted therapies.27 Several CD123CART trials for patients with relapsed/refractory AML are currently open or have recently been completed (Table 3). A phase 1 study of “biodegradable” RNA-electroporated CD123CART (NCT02623582) with short in vivo persistence conducted at the University of Pennsylvania (Penn) reported clinical tolerability without severe toxicity in 5 adults with relapsed/refractory AML, although most patients did not receive the full planned cell dosing and experienced minimal clinical responses.28 A successor phase 1 trial of lentivirus/second-generation more persistent CD123CART (NCT03766126) at Penn is currently enrolling adults aged 18 years and older. In addition, investigators at the City of Hope Medical Center recently reported promising interim results from a CD123CART phase 1 trial (NCT02159495) with responses in 3 of 6 adults with relapsed/refractory AML and receipt of subsequent allogeneic HSCT in 2 of these patients. As a precautionary measure, the City of Hope Medical Center CD123CAR construct incorporates a truncated epidermal growth factor receptor suicide switch for potential clinical CAR T-cell termination with the anti–epidermal growth factor receptor antibody cetuximab, although this strategy has not been required to date. Toxicities observed in this trial were reportedly manageable and reversible with supportive care, and prolonged on-target/off-tumor cytopenias were not observed.29 This trial is now allowing pediatric participation for children and adolescents 12 to 18 years of age with relapsed/refractory CD123+ leukemia. Finally, a first-in-child CD123CART phase 1 study will soon open at the St. Jude Children’s Research Hospital. This CD123 construct contains a synthetic CD20 sequence for potential T-cell termination via administration of the CD20 antibody rituximab if needed for clinical toxicity mitigation. CAR T-cell and antibody-based immunotherapies against numerous other target antigens have been studied in preclinical AML models (Table 4), and phase 1 trials of some of these agents are in development for adults and children with relapsed/refractory AML.22
Although the majority of current CAR T-cell products are autologous in origin, allogeneic “off-the-shelf” CAR T-cell products derived from third-party healthy donors are also under study to address potential issues of unsuccessful pheresis in heavily myelosuppressed patients, expedite treatment timing, and allow potentially more cost-effective treatment of multiple patients from one T-cell donor source. However, GVHD is a major potential issue of allogeneic cellular therapies. One strategy used with “universal” allogeneic chimeric antigen receptor T cells (UCART) has employed transcription activator–like effector nuclease gene editing technology to knock out T-cell receptor expression. Preclinical studies have shown promising activity of UCART123 against AML cells.30 Phase 1 studies of UCART123 in adults with multiply relapsed CD123+ malignancies are ongoing (NCT03190278, NCT03203369).
Exciting early results of a phase 1 trial of IL-12/IL-15/IL-18–stimulated haploidentical natural killer (NK) cells for adults with relapsed/refractory AML (NCT01998793) have also been reported by the Washington University group.31,32 Additional pilot and phase 1/2 trials are now evaluating the safety and preliminary efficacy of cytokine-stimulated NK cells in children with relapsed/refractory AML with a goal of subsequent HSCT in some patients (NCT01898793, NCT03068819). NK cell immunotherapies have potential advantages of lower reported CRS rates and shorter in vivo persistence that may limit clinical toxicity and maximize HSCT engraftment success. Finally, allogeneic transgenic T-cell receptors engineered against the human minor histocompatibility antigen 1 or other AML-associated antigens have also demonstrated impressive preclinical and clinical activity (NCT03326921) in patients with acute leukemia who relapse after HSCT; these studies are described in detail elsewhere.33,34
Currently, most early-phase cellular immunotherapy studies are intended as a bridge to allogeneic HSCT for children with potential to achieve additional remission. Concern remains that long-term AML CAR T-cell persistence could induce prolonged or permanent bone marrow aplasia, which may be mitigated by the aforementioned suicide switch approaches (eg, herpes simplex virus thymidine kinase, truncated epidermal growth factor receptor/cetuximab, CD20/rituximab, inducible caspase-9/chemical inducer of dimerization drugs).22,35 Other strategies include use of CD28 instead of 4-1BB costimulatory domains, which generally result in more potent but less persistent CAR T cells.25,36 Such approaches may limit the potential toxicity of persistent CAR T cells that may harm transplanted hematopoietic stem cells and theoretically compromise engraftment and immune reconstitution.
Clinical case 3
A 13-month-old girl with early medullary relapsed B-ALL was referred for CD19CART after poor response to salvage chemotherapy. She was initially diagnosed at 3 months of age with infant ALL harboring a KMT2A-AFF1 fusion from t(4;11) and had overt central nervous system involvement (CNS3). She had achieved initial MRD-negative remission with chemotherapy as per the COG AALL15P1 protocol (NCT02828358), but she experienced an on-therapy combined central nervous system and medullary relapse at 9 months from diagnosis. She was reinduced as per the COG AALL1331 protocol (NCT02101853) and had 3.7% end-of-reinduction flow cytometric MRD. Response evaluation at 1 month after subsequent CD19CART therapy showed 50% marrow blasts with AML immunophenotypic lineage switch with positive staining for myeloperoxidase, CD117, and CD123 and negative staining for CD19, CD22, and other B-cell antigens. She was treated with a cycle of CPX-351 (liposomal cytarabine and daunorubicin) as per the COG AAML1421 protocol (NCT02642965) and had 7% residual AML upon blood count recovery. Given her demonstrated chemoresistance and poor prognosis with early relapse and lineage switch, the girl was subsequently enrolled in a phase 1 trial of flotetuzumab, a CD123 × CD3 dual-affinity retargeting antibody (DART) immunotherapy. She achieved an MRD-negative remission after one cycle of flotetuzumab and received a second cycle before subsequent allogeneic HSCT from an HLA-matched unrelated cord blood donor.
Optimizing chemotherapy for patients in first relapse
The recent COG AAML1421 phase 1/2 trial assessed the safety and preliminary efficacy of CPX-351 (cycle 1) followed by FLAG consolidation (cycle 2) in children and adolescents with first relapsed/refractory AML.37 Thirty of the 38 treated patients (79%) had complete response (CR) or CR with incomplete platelet or hematologic recovery (CRp or Cri, respectively) as best response, and 21 of the 25 patients with CR/CRp received subsequent HSCT. Although these data represent the best CR2 rate to date in children with AML, many patients treated in the AAML1421 trial were unable to undergo subsequent HSCT and remain at high risk of second relapse. On the basis of the success of AAML1421, children with newly diagnosed AML will be randomized to standard 5-cycle chemotherapy (2 inductions + 3 intensifications) vs 2 induction cycles of CPX-351 and 3 cycles of intensification chemotherapy (with GO addition for CD33+ patients in both treatment arms) in the soon-to-open COG phase 3 trial with a goal of assessing potential improvement in outcomes.
When chemotherapy fails: can antibody-based immunotherapies overcome AML chemoresistance?
Naked monoclonal antibodies (mAbs) targeting myeloid antigens were developed as some of the first immunotherapeutic approaches for patients with AML. These therapies work by direct signaling blockade via receptors or, more commonly, via NK cell–mediated antibody-dependent cellular cytotoxicity. Initial phase 1 studies of the CD33 mAb BI 836858 and the CD123 mAb CSL360 demonstrated no benefit over conventional chemotherapy in adults with relapsed/refractory AML, and these agents are unlikely to improve outcomes when administered as monotherapy.38,39 Many mAbs have subsequently been repurposed as ADC immunotherapies with more favorable pharmacologic properties and encouraging preclinical activity, such as the CD33 ADCs lintuzumab-225Ac and vadastuximab talirine (SGN-33A) and the CD123 ADCs IMGN632 and SGN-123A. Similar to GO, these newer immunotherapies are conjugated to cytotoxic payloads40-42 or to bacterial toxins43,44 (or can be developed as radioconjugates45 ) and have potential for greater antileukemia activity. ADCs are an appealing immunotherapeutic modality for heavily pretreated and myelosuppressed patients with relapsed/refractory AML because their mechanisms of action do not require an intact immune system. ADCs can often be paired with cytotoxic chemotherapy to augment antileukemia efficacy, as was successfully done with GO in the AAML0531 trial. However, ADCs have occasionally been associated with unacceptable hepatic and infectious toxicities in patients with relapsed/refractory AML, which has led to early trial termination and cessation of further drug development in some instances.46 None of these mAbs or ADCs, with the exception of GO, have been studied in children to date, although phase 1 pediatric trials of some agents are planned.
Bispecific T cell engager (BiTE; Amgen Science, Thousand Oaks, CA) and DART immunotherapies are also under current early-phase clinical evaluation in patients with relapsed/refractory AML, inspired in part by the exciting remission reinduction rates reported in patients with relapsed/refractory B-ALL treated with the CD19 × CD3 BiTE blinatumomab that led to its recent FDA and European Medicines Agency approval.47,48 Early results from a phase 1 trial of the CD33 × CD3 BiTE AMG-330 (NCT02520427) demonstrated clinical tolerability in adults with relapsed/refractory AML with largely manageable CRS. Four of 35 treated patients achieved CR/CRi.49 Interim results from a multi-institutional phase 1 trial of the CD123 × CD3 DART flotetuzumab (NCT02152956) also reported clinical tolerability and preliminary efficacy with CRs in 5 of 27 adult patients treated at the identified maximum tolerated dose50 with plans for a successor phase 2 trial (NCT03739606). The COG Pediatric Early Phase Clinical Trial Network will evaluate the safety and preliminary antileukemia activity of flotetuzumab specifically in children and adolescents with relapsed/refractory AML in the soon-to-open ADVL1812 phase 1 trial.
Investigation of cell cycle checkpoint blockade in AML
Immune checkpoints are normal physiologic mechanisms by which the immune system downregulates itself to avoid deleterious sequelae of hyperinflammation after infection. Many cancers have taken advantage of this system by upregulating the ligands responsible for engaging these checkpoints to avoid immune surveillance. Checkpoint inhibition with PD-1/PD-L1 (eg, nivolumab, pembrolizumab) or CTLA4 inhibitors (eg, ipilimumab) has revolutionized the treatment of adults with various solid tumors, and investigators are now studying the potential efficacy of similar approaches in patients with hematologic malignancies. Results from early-phase studies of nivolumab or ipilimumab monotherapy or in combination with chemotherapy or hypomethylating agents in adults with relapsed/refractory AML have been mixed with respect to efficacy, and life-threatening inflammatory adverse events have been reported in some patients.51 Use of immune checkpoint inhibitors before or after HSCT to enhance GVL effects is potentially enticing but must be balanced with the risk of GVHD, which has been reported to be severe and even fatal in some cases. Checkpoint inhibitors have been minimally studied to date in the pediatric leukemia population. Two phase 1 trials are currently assessing the safety of combining blinatumomab and nivolumab or pembrolizumab in children with relapsed/refractory B-ALL (NCT02879695, NCT03605589). The recently opened T2016-002 phase 1 trial conducted via the Therapeutic Advances in Childhood Leukemia and Lymphoma Consortium is exploring the safety and tolerability, as well as establishing a recommended phase 2 dose, of nivolumab with 5-azacytidine in children with multiply relapsed/refractory AML (NCT03825367).
Summary
Justifiable excitement exists regarding successful integration of ADCs, bispecific antibodies, cellular therapies, and checkpoint inhibitors into the treatment of children and adolescents with AML. Testing of new agents in childhood AML has traditionally followed a laborious and slow paradigm of initial drug evaluation in adults with relapsed disease before monotherapy evaluation in children with multiply relapsed disease. This approach has theoretically limited the ability to observe potential activity in younger and healthier children vs adults with multiple comorbidities, in children at an earlier stage of leukemia relapse, or in combination with other therapies. However, the immunotherapy development climate for pediatric AML is changing, as evidenced by increased pharmaceutical collaboration with childhood cancer cooperative groups and several first-in-child clinical trials studying new agents. Some studies have also optimized the efficiency of early-phase trial designs with monotherapy “run-in” windows for safety evaluation and dose finding of new agents before subsequent combination with chemotherapy that may better maximize detection of a potential treatment efficacy signal.
As new AML immunotherapies become more commonly studied in children, it is plausible that mechanisms of immune escape and treatment resistance will emerge, as has been reported in children with B-ALL treated with CD19- or CD22-targeted immunotherapies. Combinatorial approaches with bispecific antigen targeting or immunotherapy addition to chemotherapy or with other inhibitors are thus under active development. Ideally, new immunotherapies could be used in the newly diagnosed setting for children with demonstrated chemoresistance or for those at particularly high risk of relapse with a goal of successful HSCT and/or improving EFS and OS. It is not yet clear, however, whether immunotherapies will decrease the need for subsequent HSCT in children with AML, and most current immunotherapy strategies are conservatively planned as a bridge to transplantation when clinically feasible. Ultimately, it is hoped that successful integration of new immunotherapies into the care of children with AML will ultimately allow conventional therapy reduction and decrease the significant morbidity and mortality associated with current regimens. These challenges will be difficult to navigate but are most welcome to contemplate as we strive to improve relapse-free and long-term cure rates for children with AML.
Acknowledgments
This article was authored in memory and celebration of Schyler Anna Herman.
A.J.L. was supported by the Seattle Children’s Hospital Cancer and Blood Disorders Center Research Pilot Program. S.K.T. was supported by National Institutes of Health/National Cancer Institute grants K08CA184418 and 1U01CA232486, the Rally Foundation for Childhood Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, and the St. Baldrick’s Foundation/Stand Up to Cancer Pediatric Dream Team Award. Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Authorship
Contribution: A.J.L. and S.K.T. wrote and edited the manuscript and approved the final version.
Conflict-of-interest disclosure: S.K.T. is a member of the Scientific Advisory Board for Aleta Biotherapeutics. A.J.L. declares no competing financial interests. Off-label drug use: None disclosed.
Correspondence: Sarah K. Tasian, Children’s Hospital of Philadelphia, University of Pennsylvania Perelman School of Medicine, 3501 Civic Center Blvd, CTRB 3010, Philadelphia, PA 19104; e-mail: tasians@e-mail.chop.edu.


