

Key Points
Clinically relevant imetelstat concentrations significantly inhibit CFU-MEG formation from MNCs of ET patients and reduce hTERT expression.
However, similar concentrations of imetelstat do not inhibit cytokine-induced CFU-MEG from MNCs of healthy donors.
Introduction
In most human cancer cells, indefinite replicative potential is driven by telomerase activation.1,2 Two telomerase subunits, human telomerase RNA and human telomerase–associated protein, are constitutively expressed; enzyme activity depends on a third subunit, human telomerase reverse transcriptase (hTERT).3 Approximately 90% of human cancers have increased levels of telomerase activity (TA),4 making it a potential target for cancer treatment.
Imetelstat sodium (imetelstat) is a covalently lipidated 13-mer oligonucleotide that binds to human telomerase RNA and is a potent competitive inhibitor of TA.5,6 Imetelstat demonstrated promising antitumor activity in phase 2 trials in patients with essential thrombocythemia (ET) and myelofibrosis.7,8
ET is a myeloproliferative neoplasm (MPN) characterized by sustained uncontrolled proliferation of hematopoietic precursors with spontaneous growth of myeloid and megakaryocytic colony-forming units (CFU) in vitro. Reactivation of telomerase is important for clonal expansion. hTERT expression, although not normally observed in somatic cells, has been demonstrated in megakaryocytes from patients with ET,9 refractory anemia, and chronic myeloid leukemia.10
A phase 2 study of imetelstat enrolled 18 ET patients who were refractory or intolerant to prior treatment. All patients achieved a hematologic response; 89% had a complete hematologic response, and 94% had a clinico-hematologic response.7,11 Seven of 8 patients (88%) with JAK2 V617F mutations had >50% reduction in mutant allele burden following 3 months of treatment. Mutant MPL and CALR allele burdens were also reduced (range, 15%-66%).7 In a subsequent study, among 33 patients with high-risk or intermediate-2risk myelofibrosis treated with imetelstat, 21% achieved complete or partial remission, and bone marrow fibrosis was reversed with complete remission.8
The mechanism by which inhibition of telomerase exerts such effects in ET patients is not well understood. In this study, we aimed to investigate the effects of imetelstat on megakaryocyte development from malignant and normal precursors. We assessed the potential of imetelstat to inhibit in vitro growth of megakaryocyte CFU from peripheral blood (PB) of ET patients and healthy individuals (HIs), as well as its effects on the differentiation of megakaryocytes from human cord blood (CB). In addition, we measured the expression of hTERT in ET patients compared with HIs and the reduction in hTERT in ET patients treated with imetelstat.
Methods
Mononuclear cells (MNCs) were isolated from PB of 10 ET patients and 3 HIs (Table 1). Cells were plated onto collagen in serum-free StemSpan SFEM media, with and without cytokines for colony-forming units of megakaryocytes (CFU-MEG) formation (50 ng/mL thrombopoietin [Tpo], 10 ng/mL interleukin-3 [IL-3], 10 ng/mL IL-6, 50 ng/mL stem cell factor [SCF], 3 U/mL erythropoietin; STEMCELL Technologies, Vancouver, BC, Canada). Clinically relevant concentrations (0.1, 1, or 10 μM) of imetelstat (5′-TAGGGTTAGACAA-3′) or a mismatch control (5′-TAGGTGTAAGCAA-3′) were added to the cultures at the time of plating. Cells were incubated for 10 to 12 days at 37°C with 5% CO2. CFU-MEG colonies were quantified by murine anti-human GPIIa/IIIb staining (STEMCELL Technologies) and light microscopy.
Characteristics of patients and CFU-MEG growth, with or without imetelstat
| Subject no. | Sex | Age, y* | Disease duration, d | Mutational status | Treatment* | Hb, g/dL* | Lc, × 109/L* | Plt, × 109/L* | CFU-MEG ± SD (n = 2) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spontaneous growth | Imetelstat, μM | Mismatch control oligo (10 μM) | |||||||||||
| 0.1 | 1 | 10 | |||||||||||
| ET patients | |||||||||||||
| 1 | F | 47 | 521 | JAK2 V617F | None | 14.1 | 11.3 | 700 | 18 ± 1.77 | 26 ± 1.06 | 22 ± 0.71 | 8 ± 0.35 | 28 ± 1.77 |
| 2 | F | 65 | 440 | JAK2 V617F | HU, aspirin | 11.7 | 3.6 | 342 | 33 ± 4.95 | 35 ± 1.41 | 16 ± 1.41 | 13 ± 1.41 | 37 ± 0.0 |
| 3 | F | 84 | 767 | JAK2 V617F | Radiophosphor, aspirin | 11.6 | 8 | 182 | 25 ± 2.12 | 26 ± 1.41 | 24 ± 2.83 | 11 ± 1.41 | 22 ± 1.41 |
| 4 | F | 37 | 2669 | JAK2 V617F | Aspirin | 14.7 | 13.3 | 994 | 100 ± 26.9 | 77 ± NA (n = 1) | 37 ± NA (n = 1) | 14 ± NA (n = 1) | 102 ± NA (n = 1) |
| 5 | F | 64 | 1132 | CALR type 2 | Interferon-α2a | 11.8 | 2.2 | 250 | 21 ± 0.71 | 29 ± 7.07 | 17 ± 4.95 | 21 ± 6.36 | |
| 6 | M | 74 | 1820 | Triple negative | HU, aspirin | 12.6 | 6.6 | 564 | 58 ± 10.2 | 68 ± 2.83 | 30 ± NA (n = 1) | 26 ± 14.85 | 49 ± 4.95 |
| 7 | F | 79 | 862 | CALR type 1 | HU, aspirin | 12.3 | 13.7 | 700 | 24 ± NA (n = 1) | 8 ± 1.41 | 7 ± 0.0 | 3 ± 0.71 | 22 ± NA (n = 1) |
| 8 | M | 66 | 14 | JAK2 V617F | HU, aspirin | 15.9 | 10.7 | 834 | 37 ± NA (n = 1) | 52 ± 3.54 | 18 ± 4.95 | 5 ± NA (n = 1) | 27 ± NA (n = 1) |
| 9 | M | 58 | 3716 | JAK2 V617F | HU, aspirin | 15.5 | 5.5 | 364 | 10 ± 4.24 | 8 ± 1.41 | 4 ± 0.71 | 4 ± NA (n = 1) | 11 ± 5.66 |
| 10 | M | 51 | 1 | JAK2 V617F | HU, aspirin | 16.5 | 6.9 | 677 | 37 ± NA (n = 1) | 42 ± 0.0 | 35 ± 12.73 | 18 ± 2.83 | 33 ± 7.07 |
| Subject no. | Sex | Age, y* | Disease duration, d | Mutational status | Treatment* | Hb, g/dL* | Lc, × 109/L* | Plt, × 109/L* | CFU-MEG ± SD (n = 2) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Spontaneous growth | Imetelstat, μM | Mismatch control oligo (10 μM) | |||||||||||
| 0.1 | 1 | 10 | |||||||||||
| ET patients | |||||||||||||
| 1 | F | 47 | 521 | JAK2 V617F | None | 14.1 | 11.3 | 700 | 18 ± 1.77 | 26 ± 1.06 | 22 ± 0.71 | 8 ± 0.35 | 28 ± 1.77 |
| 2 | F | 65 | 440 | JAK2 V617F | HU, aspirin | 11.7 | 3.6 | 342 | 33 ± 4.95 | 35 ± 1.41 | 16 ± 1.41 | 13 ± 1.41 | 37 ± 0.0 |
| 3 | F | 84 | 767 | JAK2 V617F | Radiophosphor, aspirin | 11.6 | 8 | 182 | 25 ± 2.12 | 26 ± 1.41 | 24 ± 2.83 | 11 ± 1.41 | 22 ± 1.41 |
| 4 | F | 37 | 2669 | JAK2 V617F | Aspirin | 14.7 | 13.3 | 994 | 100 ± 26.9 | 77 ± NA (n = 1) | 37 ± NA (n = 1) | 14 ± NA (n = 1) | 102 ± NA (n = 1) |
| 5 | F | 64 | 1132 | CALR type 2 | Interferon-α2a | 11.8 | 2.2 | 250 | 21 ± 0.71 | 29 ± 7.07 | 17 ± 4.95 | 21 ± 6.36 | |
| 6 | M | 74 | 1820 | Triple negative | HU, aspirin | 12.6 | 6.6 | 564 | 58 ± 10.2 | 68 ± 2.83 | 30 ± NA (n = 1) | 26 ± 14.85 | 49 ± 4.95 |
| 7 | F | 79 | 862 | CALR type 1 | HU, aspirin | 12.3 | 13.7 | 700 | 24 ± NA (n = 1) | 8 ± 1.41 | 7 ± 0.0 | 3 ± 0.71 | 22 ± NA (n = 1) |
| 8 | M | 66 | 14 | JAK2 V617F | HU, aspirin | 15.9 | 10.7 | 834 | 37 ± NA (n = 1) | 52 ± 3.54 | 18 ± 4.95 | 5 ± NA (n = 1) | 27 ± NA (n = 1) |
| 9 | M | 58 | 3716 | JAK2 V617F | HU, aspirin | 15.5 | 5.5 | 364 | 10 ± 4.24 | 8 ± 1.41 | 4 ± 0.71 | 4 ± NA (n = 1) | 11 ± 5.66 |
| 10 | M | 51 | 1 | JAK2 V617F | HU, aspirin | 16.5 | 6.9 | 677 | 37 ± NA (n = 1) | 42 ± 0.0 | 35 ± 12.73 | 18 ± 2.83 | 33 ± 7.07 |
| Cytokine-driven growth | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIs | |||||||||||||
| 1 | M | 51 | NA | NA | NA | 17.2 | 5 | 127 | 28 ± 4.24 | 26 ± 2.83 | 27 ± 1.41 | 24 ± 2.83 | 36 ± 4.95 |
| 2 | M | 50 | NA | NA | NA | 15.5 | 6.6 | 175 | 11 ± 2.3 | 12 ± 6.36 | 12 ± 5.66 | 19 ± 1.41 | 12 ± 1.41 |
| 3 | M | 40 | NA | NA | NA | 14.8 | 4.7 | 125 | 18 ± 0.0 | 20 ± 8.49 | 22 ± 3.54 | 14 ± 2.83 | 15 ± 2.83 |
| Cytokine-driven growth | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIs | |||||||||||||
| 1 | M | 51 | NA | NA | NA | 17.2 | 5 | 127 | 28 ± 4.24 | 26 ± 2.83 | 27 ± 1.41 | 24 ± 2.83 | 36 ± 4.95 |
| 2 | M | 50 | NA | NA | NA | 15.5 | 6.6 | 175 | 11 ± 2.3 | 12 ± 6.36 | 12 ± 5.66 | 19 ± 1.41 | 12 ± 1.41 |
| 3 | M | 40 | NA | NA | NA | 14.8 | 4.7 | 125 | 18 ± 0.0 | 20 ± 8.49 | 22 ± 3.54 | 14 ± 2.83 | 15 ± 2.83 |
F, female; Hb, hemoglobin; HU, hydroxyurea; Lc, leukocytes; M, male; NA, not applicable; Plt, platelets; SD, standard deviation.
At time of progenitor cell assay.
Five different lots of CB MNCs (Lonza; Basel, Switzerland) were enriched for CD34+ cells using negative selection with EasySep (STEMCELL Technologies). CD34+ CB cells were incubated with imetelstat at clinically relevant concentrations (0.9-15 μM) in serum-free liquid medium (StemSpan SFEM) with cytokines for megakaryocyte differentiation (1.25 ng/mL recombinant human [rh]SCF, 1.25 ng/mL rh fms-like tyrosine kinase-3 ligand, 50 ng/mL rhIL-6, and 100 ng/mL rhTpo). On day 10, the media were changed to StemSpan SFEM with 0.6 ng/mL rhSCF, 7.5 ng/mL rhIL-6, 13.5 ng/mL rhIL-9, and 30 ng/mL rhTpo. The cells were cultured for 14 days without further addition of imetelstat (in vitro half-life is estimated at ∼37 hours12 ). On days 1 through 7, 10, and 14 of culture, cells were quantified, and megakaryocyte differentiation was assessed by CD34 and CD41a using flow cytometry.
TA was measured using a modified version of the telomeric repeat amplification protocol assay by a fluorescent-labeled TS primer.13 hTERT messenger RNA (mRNA) expression was analyzed by droplet digital polymerase chain reaction, according to Dong et al,14 in PB leukocytes from 21 ET patients and 11 HIs.
Results and discussion
Imetelstat treatment resulted in significant dose-dependent suppression of spontaneously grown CFU-MEG from MNCs of ET patients (n = 10), but it had no effect on cytokine-stimulated CFU-MEG from MNCs of HIs (n = 3) (P < .0001) (Figure 1A). In comparison with CFU-MEG grown in the absence of imetelstat (= 100%), CFU-MEG from patient samples grown in 0.1 µM, 1 µM, or 10 µM imetelstat were 107% ± 8.6%, 79% ± 11.8%, and 33% ± 9.4%, respectively (mean ± standard error of the mean from 2 replicates), whereas CFU-MEG from healthy samples were 104% ± 38%, 109% ± 25%, and 112% ± 13%, respectively (Table 1). These results were confirmed using additional samples taken on the same day or on 2 separate days from 3 ET patients and from 1 HI.
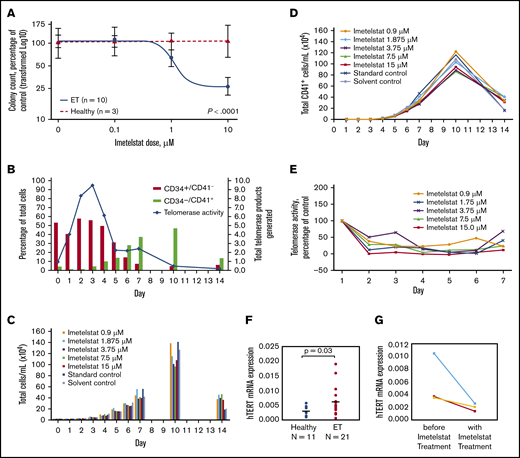
Differentiation of megakaryocytes, as indicated by increased expression of CD41+, and effects of imetelstat on CFU-MEG growth and TA, as well as hTERT expression. (A) Dose-response analysis of CFU-MEG growth from PB MNCs from patients with ET compared with HIs at different clinically relevant concentrations of imetelstat. (B) Percentage of differentiated megakaryocytes from the total number of human CD34+ CB cells incubated with cytokines for megakaryocytic development and TA. An internal control in each reaction was used to normalize the telomerase product signal. (C-D) Megakaryocytic differentiation in the presence of developmental cytokines, with and without different concentrations of imetelstat. Each panel is the result of 1 experiment. “Standard control” indicates treatment with cell culture media alone. Because imetelstat is solubilized in dimethyl sulfoxide, an additional “Solvent control” treatment with dimethyl sulfoxide alone was included. (E) TA in the presence of different concentrations of imetelstat during the development of megakaryocytes from human CB, normalized to control without imetelstat. (F) Comparison of hTERT mRNA levels in ET patients vs HIs, normalized to ABL1. (G) Reduction in hTERT mRNA levels in ET patients treated with imetelstat.
Differentiation of megakaryocytes, as indicated by increased expression of CD41+, and effects of imetelstat on CFU-MEG growth and TA, as well as hTERT expression. (A) Dose-response analysis of CFU-MEG growth from PB MNCs from patients with ET compared with HIs at different clinically relevant concentrations of imetelstat. (B) Percentage of differentiated megakaryocytes from the total number of human CD34+ CB cells incubated with cytokines for megakaryocytic development and TA. An internal control in each reaction was used to normalize the telomerase product signal. (C-D) Megakaryocytic differentiation in the presence of developmental cytokines, with and without different concentrations of imetelstat. Each panel is the result of 1 experiment. “Standard control” indicates treatment with cell culture media alone. Because imetelstat is solubilized in dimethyl sulfoxide, an additional “Solvent control” treatment with dimethyl sulfoxide alone was included. (E) TA in the presence of different concentrations of imetelstat during the development of megakaryocytes from human CB, normalized to control without imetelstat. (F) Comparison of hTERT mRNA levels in ET patients vs HIs, normalized to ABL1. (G) Reduction in hTERT mRNA levels in ET patients treated with imetelstat.
Among the 10 ET patients, 7 had a JAK2 V617F mutation, 2 had a CALR mutation (1 type 1, 1 type 2), and 1 was triple (JAK2, CALR, MPL) negative (Table 1). We observed that CFU-MEG growth was suppressed by imetelstat, regardless of driver mutation. These data are consistent with the outcomes of patients in the phase 2 study of imetelstat in ET.5 In addition, suppression of CFU-MEG growth appeared to be independent of a patient’s prior cytoreductive therapy (Table 1).
We subsequently investigated the effect of inhibiting TA with imetelstat on in vitro differentiation and proliferation of megakaryocytes from human CD34+ CB cells. In this culture system, the percentage of progenitor cells (CD34+CD41−) decreased over time as the percentage of megakaryocytes (CD34−CD41+) increased (Figure 1B). TA increased initially but was decreased as cells differentiated, and CD41 expression increased (Figure 1B). Treatment with imetelstat had a limited effect on the growth and differentiation of megakaryocytes derived from CD34+ CB cells (Figure 1C-D), despite inhibition of TA on days 1 through 7 at all concentrations of the drug (Figure 1E).
The highly significant suppressive effect of imetelstat on CFU-MEG from ET patients compared with HIs suggests a specific sensitivity of neoplastic cells to telomerase inhibition and provides evidence for a mechanism through which imetelstat exhibits clinical improvement in MPNs. An explanation for this distinct effect on neoplastic cells could be the significantly higher hTERT levels found in ET patients compared with HIs (Figure 1F). Imetelstat treatment consistently decreased hTERT levels in 3 ET patient samples (Figure 1G). Furthermore, this corroborated a recent report that imetelstat decreased hTERT levels and TA in patients with myelodysplastic syndromes and a reduction in hTERT correlated with clinical response.15 We previously reported that a reduction in TA with imetelstat treatment in vivo correlated with a decreased allele burden of the driver mutation.7 Therefore, higher hTERT levels in ET patients compared with HIs seem to play a critical role in the differential inhibition of cellular growth by imetelstat.
Mosoyan et al reported that imetelstat affected megakaryopoiesis by blocking terminal maturation of CD34+CD41+ megakaryocyte precursors, as well as inhibited the differentiation of CD34+ cells from patients with MPN to form megakaryocyte colonies.16 Our observations, in patients treated with imetelstat, confirm and extend these findings. Analogous to our data indicating an intrinsic sensitivity of neoplastic cells from ET patients to telomerase inhibition, such sensitivity may also affect neoplastic cells from patients with other MPNs or cancers.
Our results demonstrate a specificity of imetelstat for malignant megakaryocyte precursors and provide novel insights into how competitive inhibition of telomerase by imetelstat translates into clinical effects.
Acknowledgments
This study was supported in part by research funding and technical assistance from Geron Corporation (G.M.B.). Independent investigators initiated and performed this work. Editorial assistance was provided by Geron Corporation and Tamara Fink, as well as Robert Axford-Gatley (PAREXEL, Hackensack, NJ), with funding from Janssen Global Services, LLC (Raritan, NJ).
Authorship
Contribution: G.M.B. and N.G. designed the research; M.H., T.R.B., and C.B. performed the research; B.B. performed the statistical analysis; G.M.B., E.O.L., and N.G. analyzed and interpreted the data; G.M.B. and E.O.L. wrote the initial draft of the manuscript; and all authors reviewed the manuscript and approved the final version for submission.
Conflict-of-interest disclosure: G.M.B. and E.O.L. received research grant funding, as well as nonfinancial support, from Geron Corporation. N.G. and B.B. were employees of Geron Corporation at the time that the research was performed, and B.B. is a consultant for and stockholder in Geron Corporation. The remaining authors declare no competing financial interests.
The current affiliation for B.B. is Xencor, Inc., Monrovia, CA.
The current affiliation for N.G. is Amgen, Thousand Oaks, CA.
Correspondence: Gabriela M. Baerlocher, University Clinic of Hematology and Central Hematology Laboratory, Stem Cell Laboratory and Laboratory of Hematopoiesis and Molecular Genetics, Inselspital, University Hospital Bern, University of Bern, Freiburgstr 4, CH-3010 Bern, Switzerland; e-mail: gabriela.baerlocher@insel.ch.

