

Key Points
This is the first-ever demonstration of successful treatment of paroxysmal cold hemoglobinuria using the complement inhibitor eculizumab.
Introduction
We report on a 4-year-old boy who presented with acute hemolysis due to paroxysmal cold hemoglobinuria (PCH). Methylprednisolone therapy was not associated with clinical improvement, but administration of a single dose of the anticomplement antibody eculizumab was followed by rapid and durable improvement in hemolysis without toxicities. To our knowledge, this is the first report of successful use of complement blockade in the treatment of this rare but life-threatening disorder.
Case description
This 4-year-old previously healthy unvaccinated boy was in his usual state of health until 5 days prior to admission when he developed a transient, maculopapular, erythematous rash on his cheeks, trunk, and extremities. Three days prior to admission, he developed fatigue, headaches, thigh and abdominal pain, and a nonproductive cough and chills that worsened when he ate ice cream or was exposed to a cold breeze. He also developed nonbloody, nonbilious emesis, loose stools, and a fever to 40.5°C. On the day of admission, he developed jaundice and darkening of his urine. His pediatrician measured a hemoglobin level of 6.4 g/dL, prompting transfer to the emergency department, where physical examination demonstrated tachycardia, scleral icterus, and a II/VI systolic murmur. Initial diagnostic testing showed worsening of anemia to 5.6 g/dL, spherocytosis, and polychromasia on blood smear concerning for hemolysis, decreased serum haptoglobin (<6 mg/dL; reference, 36-195 mg/dL), hemoglobinuria, and elevation of nonspecific inflammatory indices (C-reactive protein, 133.8 mg/L; reference, <7.5 mg/L).
Methods
Laboratory tests drawn 1 hour later demonstrated rapid worsening of anemia (hemoglobin, 4.5 g/dL) with reticulocytopenia (25.7 × 109 cells per liter). Due to worsening tachycardia, pallor, altered mental status, and the above laboratory indices, the patient was initiated on 1 mg/kg IV methylprednisolone every 6 hours as empiric treatment of presumed autoimmune hemolytic anemia (AIHA). The polyspecific direct antiglobulin test (DAT) returned positive and was followed by a monospecific DAT that was positive for complement C3d (3+) and negative for immunoglobulin G (IgG), suggesting that the patient’s erythrocytes were coated in immunoglobulin with affinity for complement.
A cold agglutinin (CA) titration was attempted, but CA could not be detected in the serum. This finding, in combination with the presence of hemoglobinuria, suggested intravascular hemolysis and PCH as a potential diagnosis. Testing for a Donath-Landsteiner (D-L) antibody was sent. As empiric treatment of AIHA, his room was maintained at warm temperatures and he received azithromycin while awaiting results for Mycoplasma pneumoniae serologies. Other tests for infection including respiratory viral testing, testing for Epstein-Barr virus, and urine culture did not identify causative pathogens. Laboratory tests on hospital day 2 (HD2) showed worsening anemia (hemoglobin, 3.9 g/dL) with persistent reticulocytopenia. Despite transfusion with 10 mL/kg warmed packed red blood cells (RBCs; PRBCs), the patient experienced only transient increase in hemoglobin to 4.6 g/dL followed by ongoing hemolysis and a fall in hemoglobin to 3.8 g/dL.
The morning of HD3, he developed signs of shock including altered mental status, tachycardia, lactate of 3.3 mmol/L, and high oxygen extraction (PvO2 21 mmHg, SvO2 25%). Laboratory studies revealed continuing hemolysis, reticulocytopenia, and worsening anemia (hemoglobin, 3.0 g/dL; reticulocyte count, 9.5 × 109/L; lactate dehydrogenase [LDH], 1371 U/L). A second transfusion of 10 mL/kg warmed PRBCs produced a transient increase in hemoglobin to 5.2 g/dL, but on HD4 this level fell to 4.6 g/dL, suggesting methylprednisolone-refractory hemolysis.
Given ongoing refractory hemolytic anemia and shock, and with strong evidence for complement-mediated hemolysis and no clear second-line agent with demonstrated efficacy, we elected to treat the patient with a single IV infusion of 600 mg of eculizumab on HD4. Immediately following eculizumab administration, a reduction in LDH levels occurred, followed by incremental increases in the reticulocyte count, stabilization of hemoglobin levels, and no additional transfusion requirement (Figure 1). Later that day, the D-L test resulted as positive, confirming the diagnosis of PCH. Corticosteroids were maintained to control any component of extravascular hemolysis and were discontinued on HD6. On HD7, a total complement level (50% hemolytic complement) returned as <13.8 U/mL (reference, 41.7-95.1 U/mL), confirming successful complement blockade after eculizumab dosing. The patient was discharged on HD15 with a hemoglobin level of 6.6 g/dL. Follow-up testing 6 days after discharge indicated a hemoglobin level of 9.1 g/dL. Four weeks after initial diagnosis, hemoglobin had normalized to 11.3 g/dL. Diagnostic tests for infectious and toxin-mediated causes of PCH remained negative.
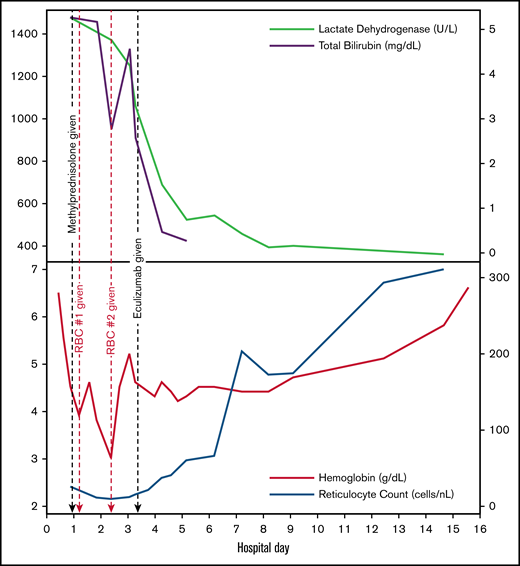
Trends in laboratory findings. Black dashed lines refer to administration of methylprednisolone (first black dashed line) and eculizumab (second black dashed line). Red dashed lines refer to administration of 10 mL/kg warmed PRBC transfusion.
Trends in laboratory findings. Black dashed lines refer to administration of methylprednisolone (first black dashed line) and eculizumab (second black dashed line). Red dashed lines refer to administration of 10 mL/kg warmed PRBC transfusion.
Given concern for an eculizumab-related increased susceptibility to encapsulated bacteria, immunization against Neisseria meningitidis was recommended but declined by the parents. Prophylaxis with penicillin VK was initiated and continued until 50% hemolytic complement levels normalized 42 days after eculizumab administration.
Results and discussion
PCH is a rare autoimmune hemolytic anemia that presents with intravascular hemolysis and hemoglobinuria. In 1904, Julius Donath and Karl Landsteiner identified the causative autoantibody in PCH.1 This polyclonal biphasic IgG antibody targets the surface carbohydrate antigen termed P on human erythrocytes. At temperatures below 37°C, the antibody binds its antigen and fixes the early complement components C1, C4, and C2. When warmed to 37°C, further activation of the classical pathway occurs, with splitting of C3 into C3a and C3b, followed by terminal complement pathway activation.2-5 This results in the formation of the membrane attack complex and fulminant intravascular hemolysis. Hence, the clinical test for the D-L antibody requires incubating 1 aliquot of patient serum with RBCs on ice for 30 minutes followed by 60 minutes at 37°C; another aliquot is exclusively incubated at 37°C for 90 minutes. Hemolysis occurring only in the cold-incubation tube indicates a positive test; exogenous complement may be added to increase sensitivity.1,2 As the autoantibody does not have CA activity, DAT is classically positive for C3 components (typically C3d) and negative for IgG, although negative DAT can occur.
Historically associated with congenital or tertiary syphilis, PCH now most commonly occurs in children following viral infection.2 It is typically a transient immune-mediated hemolysis seen 1 to 3 weeks after a viral illness, with >70% occurring after an upper respiratory tract infection. Other known precipitating factors include gastrointestinal illnesses, vaccination, hematopoietic malignancies, and autoimmune disorders. Our patient’s antecedent rash and gastrointestinal symptoms make a viral precursor likely. There is a predilection for male patients with a median age of 5 years who present with acute onset of clinically significant anemia, hemoglobinuria, jaundice, and pallor, often with hemoglobin nadiring at <6 g/dL.6,7 Reticulocytopenia as seen in our patient has been associated with PCH and occurs in the first 2 to 4 days of the disease in one-third of children.4,7,8 Reticulocytopenia likely occurs due to both physiologic lag in reticulocyte generation as well as the direct destruction of reticulocytes expressing the P antigen.9,10
Management of PCH has been primarily supportive with maintaining warm ambient temperatures to prevent further autoantibody binding, and transfusions using a blood warmer as needed. Corticosteroids have been used with unclear efficacy.8 Refractory, recurrent, and chronic PCH has been successfully treated with more intensive immunosuppressive therapies such as rituximab or IV IgG in single case reports.11,12 Eculizumab, a humanized anti-C5 monoclonal antibody that blocks the terminal complement pathway, represents an appealing potential therapeutic given efficacy in paroxysmal nocturnal hemoglobinuria, a disease in which erythrocytes are prone to complement-mediated hemolysis due to lack of membrane-bound inhibitory proteins. However, prior to this report, eculizumab use in PCH has been limited to 1 case report in which intravascular hemolysis improved substantially but ongoing anemia was attributed to extravascular hemolysis.13 The previous patient was an adult with chronic PCH and an underlying hematologic malignancy, thus not representative of postviral childhood PCH.
Eculizumab has been used in other complement-dependent AIHAs including CA disease and secondary CA syndrome; however, these are driven by IgM autoantibodies and are characterized by predominantly extravascular hemolysis rather than the intravascular hemolysis dominant in PCH.5 As such, a prospective trial in 13 patients with CA disease treated with eculizumab showed only a marginal increase in hemoglobin level but significant decreases in transfusion requirement and LDH levels, consistent with inhibition of intravascular hemolysis but poor effect on extravascular hemolysis.14 In an effort to target both intravascular and extravascular complement-mediated hemolysis, novel upstream complement inhibitors such as sutimlimab and pegcetacoplan have been developed and show early evidence of successful complement blockade, abrogation of hemolysis, and favorable side-effect profiles.15-17
In conclusion, this first-ever demonstration of rapid, complete, and sustained clinical improvement following a single dose of eculizumab in PCH presents a compelling vignette for further exploration of complement-directed therapies in this disease. Future studies are necessary to assess efficacy in a broader cohort and to determine the minimum dose and duration of therapy required to induce complement blockade while limiting the risks of opportunistic infection.18 Additionally, future studies are required to investigate the most appropriate complement inhibitor for PCH, including novel upstream complement inhibitors that would be able to target both intravascular and extravascular hemolysis.19,20
Acknowledgment
The authors acknowledge and thank the patient and family discussed herein.
Authorship
Contribution: S.A.L.-B., H.S., G.C., S.B., B.S.B., and M.S.Z. conceived this case report, drafted the initial manuscript, performed critical edits and revisions of the manuscript, and approved the final manuscript as submitted.
Conflict-of-interest disclosure: S.B. has consultancies with Apellis, Bioverative, and True North Therapeutics, as well as travel grants and lecture honoraria from Alexion and Apellis. The remaining authors declare no competing financial interests.
Correspondence: Sarah A. Lau-Braunhut, University of California, San Francisco, 550 16th St #5836, San Francisco, CA 94158; e-mail: sarah.lau-braunhut@ucsf.edu.

