Key Points
PHF6 and DNMT3A mutations define 2 distinct subgroups of MPAL with T-lineage differentiation.
Intratumoral immunophenotypic heterogeneity is independent of somatic genetic variation in MPAL.

Abstract
The genetic aberrations that drive mixed phenotype acute leukemia (MPAL) remain largely unknown, with the exception of a small subset of MPALs harboring BCR-ABL1 and MLL translocations. We performed clinicopathologic and genetic evaluation of 52 presumptive MPAL cases at Memorial Sloan Kettering Cancer Center. Only 29 out of 52 (56%) cases were confirmed to be bona fide MPAL according to the 2016 World Heath Organization classification. We identified PHF6 and DNMT3A mutations as the most common recurrent mutations in MPAL, each occurring in 6 out of 26 (23%) cases. These mutations are mutually exclusive of each other and BCR-ABL1/MLL translocations. PHF6- and DNMT3A-mutated MPAL showed marked predilection for T-lineage differentiation (5/6 PHF6 mutated, 6/6 DNMT3A mutated). PHF6-mutated MPAL occurred in a younger patient cohort compared with DNMT3A-mutated cases (median age, 27 years vs 61 years, P < .01). All 3 MPAL cases with both T- and B-lineage differentiation harbored PHF6 mutations. MPAL with T-lineage differentiation was associated with nodal or extramedullary involvement (9/15 [60%] vs 0, P = .001) and a higher relapse incidence (78% vs 22%, P = .017) compared with those without T-lineage differentiation. Sequencing studies on flow-cytometry–sorted populations demonstrated that PHF6 mutations are present in all blast compartments regardless of lineage differentiation with high variant allele frequency, implicating PHF6 as an early mutation in MPAL pathogenesis. In conclusion, PHF6 and DNMT3A mutations are the most common somatic alterations identified in MPAL and appear to define 2 distinct subgroups of MPAL with T-lineage differentiation with inferior outcomes.
Introduction
Mixed phenotype acute leukemia (MPAL) is a rare form of acute leukemia comprising 2% to 3% of all acute leukemia diagnoses.1-5 The diagnosis of MPAL is based on flow cytometric analysis of the immunophenotype, which demonstrates expression of differentiation-related antigens belonging to multiple lineages.3,6-10 Commonly, one lineage is myeloid and the other B and/or T lymphoid. Whether lineage differentiation determines clinical presentation and outcomes is unclear.3,11-13 With the exception of the subset of cases harboring BCR-ABL1 or MLL translocations, the recurrent genetic aberrations of MPAL are not well elucidated. Recent studies have revealed abnormal karyotypes, including chromosomal translocations, in the majority of MPAL cases and also shed light on the mutational landscape of MPAL.14-23 In particular, DNMT3A is frequently mutated in MPAL and often associated with a T/myeloid phenotype.14,19,23 Many studies of MPAL have included a proportion of cases (ranging from at least 10%-30%) with monosomal or complex karyotypes involving chromosomes 5 and/or 7 that would be classified as acute myeloid leukemia (AML) with myelodysplasia-related changes (MRC) according to the current World Health Organization (WHO) definition.4,12,13,15,19,23-26 The inclusion confounds previous results and the reported association between MPAL and adverse clinical outcome. We have recently reported overall good outcomes in a well-defined cohort of MPAL patients undergoing allogeneic transplant, in contrast to prior studies, which were confounded by the inclusion of secondary AML cases.27 Given the rarity of the disease and the evolving definition of this entity, the mutational repertoire of MPAL has not been fully elucidated. In addition, the mutations associated with specific lineage differentiation and the enrichment of mutations in specific cellular compartments have not been studied in MPAL. Addressing these questions may provide new insight into the pathogenesis and inform potential therapeutic targets in specific MPAL subtypes.
The plant homeodomain finger 6 gene (PHF6) is located at Xq26–27 and encodes a zinc-finger–containing protein.28 Previous studies have demonstrated that PHF6 interacts with chromatin through its PHD-like zinc-finger domains,28,29 and with multiple transcription factors.30-33 While it has been postulated to act as a tumor suppressor,34 the precise mechanisms of PHF6 mutations in leukemic transformation remain unknown. Somatic loss-of-function PHF6 mutations are frequent in T-lymphoblastic lymphoma/leukemia (T-ALL; up to 30%) and are infrequently seen in AML (∼3%), where they are associated with AML-MRC.34-39 PHF6 mutations have also been reported in rare MPAL patients.14,27 Here, we report the clinicopathologic analysis and genomic landscape in a stringently confirmed cohort of MPAL patients from Memorial Sloan Kettering Cancer Center (MSKCC).
Methods
Patients
A cohort of 58 patients with a presumptive diagnosis of MPAL from January 2010 to December 2017 was initially obtained by searching the MSKCC patient database. Posttransplant outcomes of a subset of the patients were previously reported.27 The clinical, morphologic, immunophenotypic, and cytogenetic/molecular results were independently re-reviewed by 2 hematopathologists (W.X. and M.R.). Six patients had no evaluable immunophenotypic data and thus were excluded. We applied the new diagnostic criteria of MPAL according to the WHO 2016 classification and excluded AML with recurrent cytogenetic abnormalities, AML-MRC, and therapy-related AML (t-AML). MPAL cases with B-lymphoblastic leukemia/lymphoma (B-ALL) phenotype and isolated myeloperoxidase expression (iso-MPO) were revised to B-ALL with iso-MPO and excluded. This study was approved by the institutional review board (IRB) at MSKCC, and all patients provided informed consent.
Chromosome, FISH, and microarray analysis
Conventional chromosome analysis of bone marrow, peripheral blood, or fresh tissue was performed following standard procedures with overnight short-term culturing without mitogen. At least 20 metaphase cells were analyzed for a complete chromosome study. The chromosome abnormalities were recorded as per the International System for Human Cytogenetic Nomenclature (2016). In most cases, fluorescence in situ hybridization (FISH) analysis was performed for recurring chromosome abnormalities in AML and ALL, including t(9;22), MLL, and t(12;21). In a few cases with inadequate chromosome analysis, extensive FISH tests for t(8;21), inv(16), t(6;9), and IKZF1 were also performed. All FISH probes were from Abbott Molecular (Des Plaines, IL), and quality and performance were validated in the laboratory. Genomic microarray tests (Cytoscan chip, Affymetrix) were performed in 13 patients with available bone marrow and or peripheral blood specimens for genomic DNA extraction. A total of 250 ng DNA was digested with Nsp, followed by ligation and amplification. After purification and quantitation, the polymerase chain reaction products were fragmented, followed by labeling and hybridization. The chips were washed, stained, and scanned. The CYCHP files generated from the raw CEL files of each sample was analyzed using Chromosome Analysis Suite (ChAS 3.2, Affymetrix), with manual reviewing of recurring cancer genes, particularly leukemia-related genes. For copy-neutral loss of heterozygosity (CN-LOH), a cutoff call of 10 Mb was used.
Flow cytometry and cell sorting
Multiparameter flow cytometry was performed on bone marrow aspirates at diagnosis and/or relapse. Briefly, up to 1.5 million cells from freshly drawn bone marrow aspirate were stained with 4 to 6 10-“color” panels (supplemental Table 1), washed, and acquired on a Canto-10 cytometer (BD Biosciences, San Jose, CA). The results were analyzed with custom Woodlist software (generous gift of B. L. Wood, University of Washington). Cryopreserved bone marrow aspirate specimens collected at diagnosis from our institutional, IRB-approved biospecimen bank were sorted into myeloid blasts, T lymphoblasts and/or B lymphoblasts, and natural killer cells (as somatic controls) using FACS-Aria Fusion cell sorter (BD Biosciences).
Sequencing studies
Bone marrow samples obtained were submitted to a 28-gene amplicon capture-based next-generation sequencing (NGS) assay (RainDance) or a larger 400-gene amplicon capture-based NGS assay (FoundationOne Heme) as previously described.40,41 In addition, an RNA-based targeted sequencing assay (Archer FusionPlex Heme Kit) that detects and identifies fusions of 87 genes associated with hematological malignancies was performed. Variants were detected through our clinical workflow, which includes sequencing by Foundation Medicine and Raindance custom panels. These putative variants were further analyzed through a custom annotation pipeline, including VEP, Vagrent, Exac, and Gnomad, to evaluate the likely role of specific mutations in MPAL pathogenesis (supplemental Methods). Variants were categorized as oncogenic, likely, or unknown. Oncogenic and likely variants were reported in Figure 2.
Sorted cells of specific populations from specific MPAL patients were submitted for MSK IMPACT testing.42 For MSK IMPACT data analysis, short insert paired-end reads were aligned to the GRCh37 reference human genome with 1000 Genomes decoy contigs using BWA-mem43 (supplemental Methods). After alignment, we obtained an average of 500× coverage per sample. Single base substitutions were called using CaVEMan as described previously.44 Small somatic insertions and deletions (indels) were identified using a modified version of Pindel (https://github.com/cancerit/cgpPindel).45 The entire coding sequences of PHF6, WT1, RUNX1, and DNMT3A are covered by all 3 NGS assays (RainDance, FoundationOne Heme, and MSK IMPACT).
RNA-sequencing studies were also performed on sorted cells. Fusion discovery was performed using STAR-Fusion (Version 1.1.0), SOAPfuse (Version 1.27), and FusionCatcher (Version 0.99.7c). Fusions were merged and filtered by FusionCatcher databases (healthy, conjoing, hpa, banned, paralogs, 1000 Genomes, readthrough). Confident fusions are fusions called by ≥2 different callers and supported by ≥8 spanning reads.
Statistics
The Wilcoxon rank-sum and Kruskal-Wallis tests were used to compare continuous clinical factors by lineage differentiation and mutational subgroups. The Fisher’s exact test was used to compare categorical factors. Cumulative incidence functions and Gray’s test were used to compare the incidence of relapse by subgroups, while Kaplan-Meier curves and the log-rank test were used to compare overall survival and relapse-free survival. Overall survival was defined as the time from diagnosis to death or last follow-up. Relapse-free survival was defined as the time from achievement of a complete response to relapse, death, or last follow-up. All analyses were conducted using R v3.3.346
Results
Only a subset of acute leukemia with a mixed phenotype is bona fide MPAL
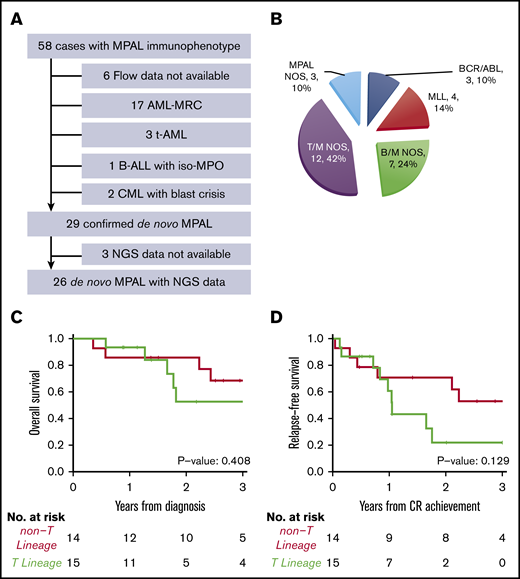
As cross-lineage antigen expression is common in AML-MRC, t-AML, and chronic myeloid leukemia in blast crisis, we carefully reviewed the clinical history, morphologic, cytogenetic, and molecular findings of 52 patients who showed mixed phenotype by flow cytometry. A total of 23 cases were excluded due to a revised diagnosis of AML-MRC (n = 17), and t-AML (n = 3), chronic myeloid leukemia with blast crisis (n = 2), B-ALL with iso-MPO (n = 1) (Figure 1A). Only 29 out of 52 cases(56%) were confirmed as bona fide MPAL according to the 2016 WHO classification.
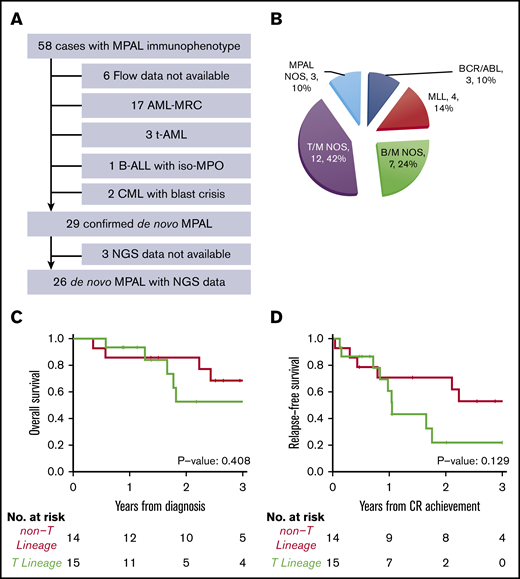
Diagnosis and subtypes of de novo MPAL. (A) Inclusion and exclusion criteria of our MPAL cohort. (B) WHO classification of our MPAL cohort. (C) Overall survival of MPAL. (D) Relapse-free survival of MPAL.
Diagnosis and subtypes of de novo MPAL. (A) Inclusion and exclusion criteria of our MPAL cohort. (B) WHO classification of our MPAL cohort. (C) Overall survival of MPAL. (D) Relapse-free survival of MPAL.
None of the 29 patients had a prior history of myeloid neoplasms. A total of 22 patients had abnormal cytogenetics (2 detected only by microarray), 6 patients had a normal karyotype, and 1 had normal FISH results. No patient had monosomies or deletions involving chromosomes 5 or 7 commonly seen in AML-MRC or t-AML (Table 1). Three patients had a prior history of malignancy (Table 2). One patient had classical Hodgkin lymphoma 8 years ago and now presented as T/B MPAL; this patient had no t-AML–defining cytogenetic abnormalities and no myeloid differentiation. Two other patients had stage I lung cancer and melanoma, respectively, and only received surgery as treatment of their solid tumor. Another patient was treated for HIV. Therefore, a diagnosis of de novo MPAL was established over AML-MRC/t-AML in these 29 patients.
MPAL with T-lineage differentiation has frequent nodal involvement and higher relapse incidence
Among the 29 confirmed MPAL patients, 14 had B/M phenotype, 12 T/M, 1 T/B, and 2 T/B/M (Figure 1B), a distribution similar to previous studies.3,4,7,15,18 A total of 4 patients (14%) had MLL translocations, and 3 (10%) had BCR-ABL1 translocations, which represent 2 distinct genetically defined subgroups. In addition, t(12;21) with ETV6-RUNX1 fusion was seen in a pediatric patient, and t(10;11) with PICALM-MLLT10 fusion was observed in 2 adult patients, respectively. Notably, the 2 patients with t(10;11) showed T-lineage differentiation. We grouped MPAL according to the presence (n = 15) or absence (n = 14) of T-lineage differentiation. The 2 groups had similar clinical characteristics, including age, sex, complete blood count (CBC), frequencies of abnormal karyotype (including complex karyotype), induction therapy, achieving complete remission (CR) after induction, and receiving hematopoietic stem cell transplant (HSCT) (Tables 1 and 2). MPAL with T-lineage differentiation had more frequent extramedullary involvement, including lymph nodes and skin (9/15 [60%] vs 0, Fisher’s exact test P < .001). In contrast to the previous studies that failed to show any differences in outcomes,3,11 we observed a higher relapse incidence (2-year cumulative incidence: 78% [95% confidence interval (CI), 32%-95%] vs 22% [95% CI, 5%-47%], Gray’s test P = .017) and a trend toward inferior progression-free survival (2-year relapse-free survival: 30% [95% CI, 12%-76%] vs 71% [95% CI, 51%-99%]; log-rank P = .129) in MPAL with T-lineage differentiation, although the overall survival was not significantly different (log-rank P = .408; Figure 1C-D). The median follow-up among survivors was 2.6 years (0.6-6.1 years).
Genomic landscape of MPAL
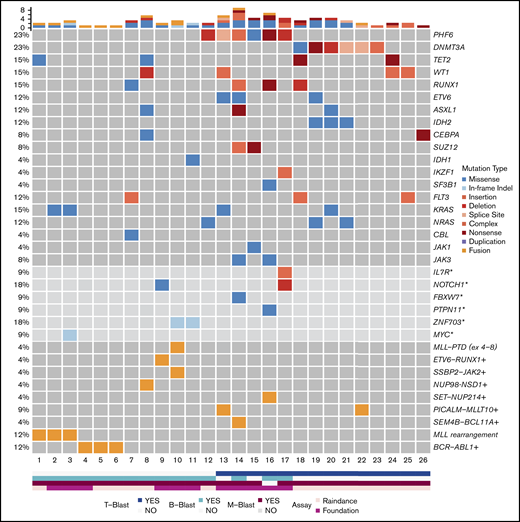
Mutational results were obtained by targeted sequencing studies in 26 out of 29 cases (Figure 2). The 3 cases without mutational data included 1 MPAL with MLL translocation, 1 with B/M NOS, and 1 with T/M NOS. A 28-gene panel was performed on 26 cases; 11 of these cases were also studied with a larger 400-gene panel. Among the 28 genes covered by both panels, the most commonly mutated genes were PHF6 (6/26, 23%), DNMT3A (6/26, 23%), TET2 (4/26, 15%), WT1 (4/26, 15%), RUNX1 (4/26, 15%), KRAS (4/26, 15%), FLT3 (3/26, 12%; 2 ITD and 1 TKD), ETV6 (3/26, 12%), ASXL1 (3/26, 12%), IDH2 (3/26, 12%), and NRAS (3/26, 12%). Other genes with recurrent mutations included CEBPA, SUZ12, and JAK3, occurring in 2 cases (8%) each. Mutations in IDH1, IKZF1, SF3B1, CBL, and JAK1 were detected in only 1 case (4%) each. Among the genes only covered by the FoundationOne Heme panel, we identified mutations in NOTCH1 (2/11, 18%), ZNF703 (2/11, 18%), IL-7R (1/11, 9%), FBXW7 (1/11, 9%), PTPN11 (1/11, 9%), and MYC (1/11, 9%). Within the genes shared by both panels, the mutational burden was higher in patients with T-lineage differentiation (41 mutations in 14 patients [2.9 mutations per patient] as compared with 13 mutations in 12 patients without T-lineage differentiation [1.1 mutations per patient]). Similar to a recent published study,47 mutually exclusive alterations in WT1, ETV6, RUNX1, and CEBPA were observed in 7 out of 10 cases. In contrast to the high frequency (nearly 50%) in pediatric MPAL with T/M, WT1 mutations were only observed in 2 out of 12 adult cases (16.7%). Notably, TP53 or NPM1 mutations were not detected in any patient in our cohort.
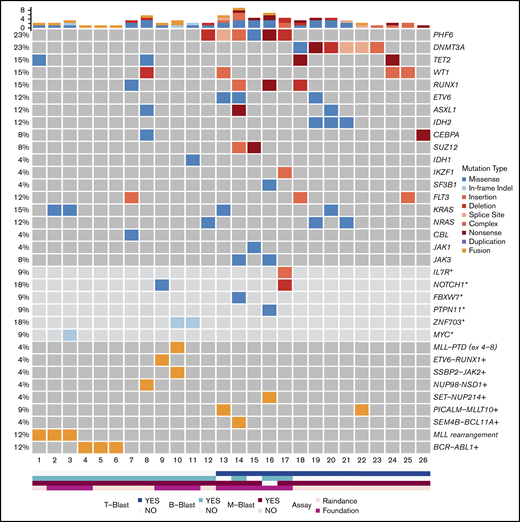
Mutational landscape of MPAL. Asterisks denote genes covered by the FoundationOne Heme panel, but not the RainDance panel, and the frequencies of these mutations in 11 patients were calculated. Plus signs denote fusions detected by Archer, RNA-seq, and/or FoundationOne Heme panels, and the frequencies of these fusions in 23 patients were calculated.
Mutational landscape of MPAL. Asterisks denote genes covered by the FoundationOne Heme panel, but not the RainDance panel, and the frequencies of these mutations in 11 patients were calculated. Plus signs denote fusions detected by Archer, RNA-seq, and/or FoundationOne Heme panels, and the frequencies of these fusions in 23 patients were calculated.
Fusion genes were examined in 23 patients (Figure 2; Table 1; supplemental Table 1). In addition to BCR-ABL1 and MLL rearrangements, 6 fusions were detected in 7 cases, including PICALM-MLLT10 (2), SEM4B-BCL11A (1), ETV6-RUNX1 (1), SSBP2-JAK2 (1), SET-NUP214(1), and NUP98-NSD1 (1).
Microarray studies were performed in 14 patients (Table 1). The concordance of array results with those of cytogenetics and/or FISH tests was excellent, generally matching the interpretation in chromosome reports. Microarray tests detected various additional genomic imbalances (mainly focal deletions) in 12 of 14 patients. In 2 remaining patients, one had a normal karyotype and the other had t(14;19). Microarray tests detected several deletions involving the genes that are known in implication of B-ALL. Two patients had t(9;22); a deletion of the EBF1 gene (5q33.2) and IKZF1 (7p12.1) was detected in one patient, whereas a deletion of RUNX1 and ERG on 21q and a deletion of MYB (6q2.3) were observed in the other patient. In one patient with t(4;11), a homozygous deletion of CDKN2A/B, along with a low level gain of chromosomes 6 and 20, was detected. In a case with normal karyotype, a deletion of PIM1 at 6p21, and CN-LOH of 21q involving the RUNX1 gene, were detected. In another case with a constitutional t(5;11), deletion of 4p and gain of chromosome 12 were revealed. In addition, genomic array tests also revealed CN-LOH in various regions, including PAX5, FLT3, RB, and GATA3 in one case, DNMT3A in one DNMT3A mutated case, and CDKN2A/B in one case. ETV6 deletions were detected in 3 out of 9 MPALs with T-lineage differentiation. These deletions and CN-LOH of various genes are indicative of the cooperation of these genes in leukemogenesis and disease progress.
PHF6 and DNMT3A mutations are the most common somatic mutations in MPAL and are mutually exclusive
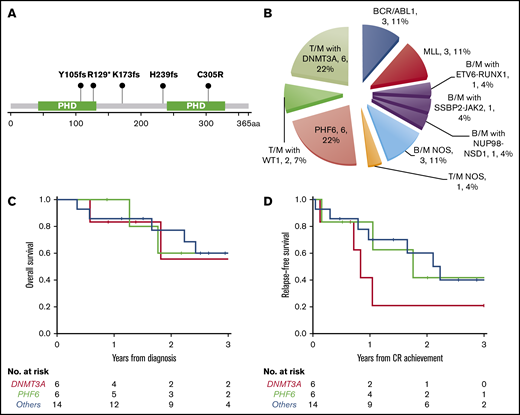
PHF6 and DNMT3A mutations were the most common alterations detected (in 6 patients each) and were mutually exclusive. The variant allele frequencies (VAFs) of PHF6 and DNMT3A mutations were higher or equal to that of other concurrent mutations in 5 cases, suggesting early clonal events (supplemental mutation data). The PHF6 mutations included 3 frameshift, 1 nonsense, 1 missense, and 1 splicing site mutation (Figure 3A). The majority of DNMT3A mutations were not at the R882 hotspot, which is most commonly seen in myeloid neoplasms (supplemental mutation data), such that the DNMT3A mutational spectrum was similar to T-ALL.48 Taken together, 46% (12/26) of the MPALs and 79% (11/14) of the MPALs with T-lineage differentiation had either PHF6 or DNMT3A mutations. These mutations were also mutually exclusive of BCR/ABL1 or MLL (Figure 2). Mutations in epigenetic regulators (WT1, RUNX1, ETV6, and ASXL1) were common in MPAL with T/M phenotype, with no major difference seen between PHF6-mutated vs DNMT3A-mutated cases, other than the absence of TET2 mutations in PHF6-mutated cases. SUZ12 (2), JAK1 (1) and JAK3 (2) mutations were only seen in PHF6-mutated cases, while IDH2 mutations (3) were exclusively detected in DNMT3A-mutated cases. Among 3 MPAL patients with T-lineage differentiation but with no PHF6 or DNMT3A mutations, 2 had WT1 mutations.
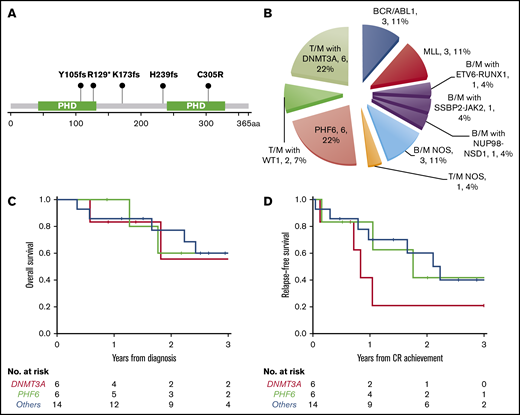
New molecular subtypes of de novo MPAL. (A) Graphic demonstration of mutant alleles of PHF6 gene (1 patient had splicing site mutation not illustrated here). (B) New MPAL classification with incorporation of our genetic discoveries. (C-D) Overall survival (C) and relapse-free survival (D) of PHF6 mutated vs DNMT3A unmutated vs others without PHF6 or DNMT3A mutations.
New molecular subtypes of de novo MPAL. (A) Graphic demonstration of mutant alleles of PHF6 gene (1 patient had splicing site mutation not illustrated here). (B) New MPAL classification with incorporation of our genetic discoveries. (C-D) Overall survival (C) and relapse-free survival (D) of PHF6 mutated vs DNMT3A unmutated vs others without PHF6 or DNMT3A mutations.
PHF6 and DNMT3A mutations appear to define 2 distinct subgroups of MPAL with T-lineage differentiation
We further compared the clinical features of PHF6- or DNMT3A-mutated MPAL to the ones without PHF6 or DNMT3A mutations (Table 3). Patients with PHF6-mutated MPAL were significantly younger than those with DNMT3A-mutated MPAL (P = .016). Both PHF6- and DNMT3A-mutated MPAL had more frequent nodal involvement than cases without either mutation (overall P < .001). There were significant differences in both hemoglobin and platelet levels across the 3 groups (P = .014 and 0.037, respectively), with the highest values of both counts being observed in the PHF6 cohort. Five of the 6 PHF6-mutated patients and all 6 DNMT3A-mutated cases showed T-lineage differentiation, in contrast to MPAL with BCR/ABL1 or MLL translocations, which were characterized by B/M differentiation (Figure 3B; Table 3; supplemental Figure 1). Of note, all 3 cases of MPAL NOS with rare T/B/M and T/B phenotypes had PHF6 mutations. All 3 groups obtained similar CR rates after induction. The majority of patients in all 3 groups underwent allo-HSCT (4/6 vs 3/6 vs 10/14). A comparison of outcomes for these patients is limited by the sample size. There appeared to be no difference in overall survival across the 3 cohorts (Figure 3C), while there was a trend toward inferior progression-free survival among DNMT3A-mutated MPAL patients; however, the sample size precludes any formal comparisons. The cumulative incidence of relapse appeared to be highest at 2 years for DNMT3A- and PHF6-mutated patients (79% and 58%, respectively) compared with the cases without PHF6 or DNMT3A mutations (33%).
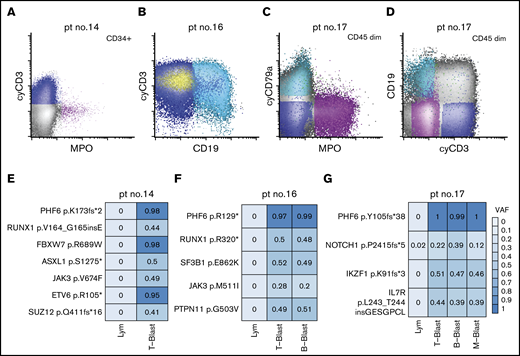
PHF6 mutations are present in all the blast compartments regardless of lineage
Given mutations in DNMT3A have been shown to be clonal and acquired in hematopoietic stem cells with multilineage differentiation (HSCs),14,49-51 we next focused on the characterization of the PHF6 mutations in MPAL. To investigate the cellular origin and clonal complexity of PHF6 mutations, we obtained viably cryopreserved bone marrow aspirate specimens of 3 PHF6-mutated patients (patients 14, 16, and 17) collected at diagnosis from our institutional, IRB-approved biospecimen bank. We successfully purified lineage-defined blast populations (ie, T-, B-, and myeloid blasts) by flow cytometry cell sorting (Figure 4A-D). In patient 14, the yield of clearly myeloid blasts was too low for downstream sequencing studies. Nevertheless, NGS of all coding regions in 400 genes implicated in hematologic malignancies (MSK IMPACT) was successfully performed on other sorted blast populations (Figure 3E-G). We confirmed the mutational profiles previously identified by our 28-gene targeted-panel NGS assay (Figure 2). All the identified mutations were present in every sorted blast population, albeit with varying frequencies. Moreover, the VAFs of PHF6 mutations were nearly 100% in every neoplastic population, indicating that PHF6 is an early pathogenic mutation during MPAL leukomogenesis. In contrast, JAK3 p. M511I in patient 16 and NOTCH1 p.P2415fs*5 in patient 17 had lower VAF values, suggesting subclonal or lineage-biased evolution.
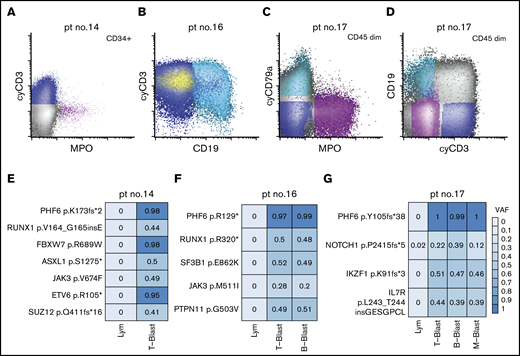
Distribution of somatic mutations in immunophenotypically distinct subpopulations in de novo MPAL. (A) An example of MPAL with T/B/M differentiation (patient 14 in Figure 2; B-blast component is not shown). (B) An example of MPAL with T/B differentiation (patient 16 in Figure 2). (C-D) An example of T/B/M MPAL by flow cytometry (patient 17). (E-G) VAFs of various mutated genes in sorted blast populations (E, patient 14; F, patient 16; G, patient 17). Lym, sorted natural killer cell population serving as germline reference.
Distribution of somatic mutations in immunophenotypically distinct subpopulations in de novo MPAL. (A) An example of MPAL with T/B/M differentiation (patient 14 in Figure 2; B-blast component is not shown). (B) An example of MPAL with T/B differentiation (patient 16 in Figure 2). (C-D) An example of T/B/M MPAL by flow cytometry (patient 17). (E-G) VAFs of various mutated genes in sorted blast populations (E, patient 14; F, patient 16; G, patient 17). Lym, sorted natural killer cell population serving as germline reference.
We next examined the differential gene expression between sorted populations from PHF6-mutated MPAL cases. Using a gene set differentially expressed in myeloid, T, and B cells,52 we showed that myeloid blasts had a higher expression level of myeloid-enriched genes (such as CSF3R, TNFSF13B, and RNASE2), while B blasts had upregulated expression of B-cell–enriched genes (CD19, BLK, and BANK1). Interestingly, T-cell–enriched genes (CD3D, CD3E, CD3G, and TBXAS1) were upregulated in both T and B blasts (supplemental Figure 3), and the CD79A/B expression level was not different among all 3 populations.
Discussion
Although mutations in DNMT3A, TET2, RUNX1, WT1, and various rare chromosomal translocations have been previously identified in MPAL,14-22 recurrent molecular alterations specific to MPAL have remained elusive. Our study demonstrates recurrent PHF6 mutations in a substantial proportion (23%) of MPAL patients. This makes PHF6 mutations as frequent as the combined proportion of BCR-ABL1 and MLL translocated MPAL patients. The relatively small number of patients with the rare disease in this study makes an estimation of the precise frequency of PHF6 -mutated MPAL difficult. Regardless, our results indicate that PHF6 mutations define a unique subset of MPAL, mostly with T-lineage phenotype presenting at young age, similar to the observations from a recent comprehensive study of pediatric MPAL.47 Notably, all MPAL patients with the extremely rare T/B and T/B/myeloid phenotypes had PHF6 mutations, suggesting PHF6 mutations might be overrepresented in this particular subset of MPAL, which is further supported by a recent collaborative study.53 The reported lower frequencies of PHF6 mutations by other studies are likely due to inclusion of MPAL mimics, bias of case selection, or the difference in PHF6 gene coverage.14,19,23 We also confirmed frequent DNMT3A mutations in MPAL14,19,23 and further showed that these cases had unique immunophenotypic and clinical features, including specific T/M differentiation,14 old age, frequent nodal involvement, and inferior outcome. PHF6 and DNMT3A mutations and BCR-ABL1 and MLL translocations together comprise ∼70% of MPALs. In addition, fusion genes were identified in 3 B/M MPAL cases, including ETV6-RUNX1, a well-known recurrent fusion gene in B-ALL; NUP98-NSD1, both reported in MPAL3,23,54 ; and a SSBP2-JAK2 fusion, which has only been previously reported in one B-ALL case.55 The 3 patients with these fusion genes had 2 distinct blast populations (one consisting of B-lymphoblasts and the other of myeloid blasts), excluding the diagnosis of B-ALL. The frequently detected ZNF384 rearrangements in pediatric MPAL with B/M phenotype were notably absent from adult patients in our series.23,47 The other 3 fusions (SET-NUP214, PICALM-MLLT10, and SEM4B-BCL11A) were seen in MPAL patients with T/M phenotype. Although the former 2 fusions have been reported in T-ALL and MPAL,36,56-62 SEM4B-BCL11A is a novel fusion with yet-to-be demonstrated function. More efforts are needed to investigate additional recurrent molecular alterations in MPAL.
PHF6 mutations have been identified in up to 25% of the pediatric early T precursor (ETP) ALL cases, where PHF6 was found to be comutated with NRAS, FLT3, WT1, EZH2, and DNM2 and less frequently with RUNX1 and SUZ12.63 While we considered that PHF6-mutated MPAL may represent a unique subset of ETP-ALL, PHF6-mutated T/M MPAL cases all had distinct MPO-positive myeloid blast populations in addition to expression of other myeloid antigens. This expression pattern excludes the diagnosis of ETP-ALL.10 MPAL cases did share some mutations with ETP-ALL (RUNX1 and SUZ12), but not the more frequent ones seen in ETP-ALL (FLT3, WT1, and EZH2), suggesting both similarities and differences between PHF6-mutated MPAL and ETP-ALL. It is not unreasonable to speculate that these 2 entities represent a biological continuum that might also be related to the newly proposed acute myeloid/T-lymphoblastic leukemia.64 DNMT3A-mutated MPAL also shows a T/M phenotype with mutations frequently seen in AML, although these patients are significantly older than patients with PHF6-mutated cases.14
Previous studies have suggested that PHF6 is critical in lineage determination, particularly between T and B lineages. CRISPR-Cas9–mediated knockout of PHF6 in B-ALL cells severely compromised the survival of B-ALL cells; mice transplanted with PHF6-deficient B-ALL cells by contrast developed lymphomas with T-ALL phenotype.29 At a gene expression level, PHF6-deficient cells have reduced expression of genes associated with B-cell development and function and enrichment of gene sets associated with T-lineage signal transduction and function. Therefore, PHF6 likely functions as a lineage-determining gene by promoting B-lineage differentiation while suppressing T-lineage differentiation, which would explain why frequent loss-of-function PHF6 mutations are found in T-ALL,34,63 but not B-ALL (1/44; www.c-BioPortal.org). Frequent T-lineage differentiation is also observed in our PHF6 mutated MPAL cohort, which further supports the functional role of PHF6.
Our sequencing data on sorted blast populations from MPAL suggest origin from early, multilineage stem/progenitors in this disease. Even though various blast populations with distinct expression of defining T, B, and/or M markers are clearly present and can be reliably purified, no definitive lineage-specific mutations are identified. Furthermore, as shown by the near-universal presence of PHF6 and other mutations in all blast compartments in our study, these mutations in MPAL likely occur at an earlier stage in more primitive progenitors such as HSCs as compared with presumptive pro/pre-T cells in T-ALL, which may prime progenitors for lineage infidelity. Distinct methylation profiles are observed between leukemia subtypes such as MPAL with B/M, MPAL with T/M, ALL, and AML, but not between sorted MPAL subclones, indicating that cytosine methylation does not drive immunophenotypic heterogeneity in MPAL.23,47 Differential gene expression was observed between sorted blast populations, although a more extensive approach including single-cell RNA-sequencing studies will be required to further delineate how differential gene expression correlates intratumoral heterogeneity in MPAL.
DNMT3A mutations are commonly found in myeloid neoplasms as well as in clonal hematopoiesis and are thought to originate from HSCs.49-51,65-67 DNMT3A also functions as a tumor suppressor in T-ALL and DNMT3A mutations are frequent in ETP-ALL.48,68 Whether DNMT3A mutations drive the pathogenesis of MPAL remains to be clarified. DNMT3A-mutated MPAL commonly acquires comutations (IDH2 and RAS) common in AML, suggesting that this subset of MPAL shares pathogenetic similarity to AML. However, the spectrum of DNMT3A mutations in MPAL is different from that of myeloid neoplasms but similar to that of adult ETP-ALL.48 In addition, the nodal involvement frequently observed in these patients is rarely seen in AML. In fact, nodal involvement is very common in MPAL with T-lineage differentiation, a phenomenon not carefully examined before. Furthermore, our data also suggest, in contrast to prior studies,3,11,13,24 worse outcome in MPAL with T-lineage differentiation.12 The discrepancies might be attributed to the differences in treatment modalities and the contamination of some prior studies by AML-MRC and t-AML.3,11,13,19,24
The limitations of this study include the relative small number of cases and targeted approaches in sequencing efforts. However, the MPAL cases in this study were stringently confirmed by exclusion of any other MPAL mimics. Taken together, we identified PHF6 and DNMT3A mutations as the most common somatic alterations in MPAL that define 2 distinct subgroups of MPAL with T-lineage differentiation and appear to lead to inferior outcomes. We also demonstrated that intratumoral immunophenotypic heterogeneity is independent of somatic genetic mutations. The pathogenesis of these mutations in MPAL needs to be addressed in subsequent genomic and functional studies.
Note added in proof
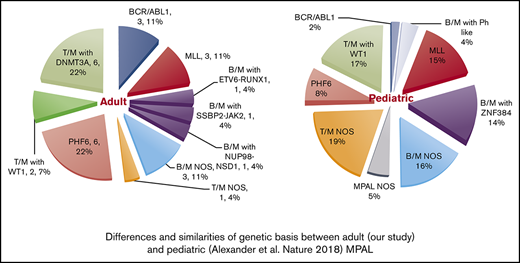
While this article was under review, 3 other MPAL studies were published that further elucidate the complexity of genetic basis in MPAL.23,47,53 In pediatric MPAL, ZNF384 rearrangements are present in nearly half of B/M cases, and WT1 mutations are present in half of the T/M cases. Both aberrations are absent or infrequent in adult MPAL. In contrast, DNMT3A mutations, frequently detected in adult MPAL with T/M, are not observed in pediatric counterparts. PHF6 mutations are present in both groups and are likely associated with T/B phenotype. BCR-ABL rearrangements are rare in pediatric MPAL. Therefore, there is marked difference in genetic basis between pediatric and adult MPAL (supplemental Figure 1). Superiority of ALL-type treatment was observed in pediatric MPAL [Hrusak O, de Haas V, Stancikova J, et al. Blood. 2018;132(3):264-276], but whether this can be translated to adult MPAL is unclear.
Presented in an abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2017.
Acknowledgments
This study was supported by the Center for Hematologic Malignancies at MSKCC and in part through the National Institutes of Health, National Cancer Institute (Cancer Center Support Grant P30 CA008748). W.X. is supported by a startup fund from Department of Pathology at MSKCC. A.H. is supported by The Swedish Cancer Society and Pediatric Cancer Society, Lion’s research fund in Skåne, The Swedish Society of Physicians, and Georg Danielsson’s fund for blood disease.
Authorship
Contribution: W.X., R.L.L., and M.R. conceived the study, collected and analyzed the data, and wrote the manuscript; M.B., M.L., J.S.M., N.F., and M.A.P. annotated the sequencing data; E.P. provided analytical oversight of the sequencing data; F.P., A.H., C.F., N.L., J.P., J.B., R.C., and M.E.A. collected data; Q.G. helped cell sorting; B.M.G., B.S., S.G., and R.R. provided critical clinical information; S.D. performed statistical analysis; Y.Z. reviewed cytogenetic data; and all the authors approved the final version of the manuscript.
Conflict-of-interest disclosure: R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Loxo, Imago, C4 Therapeutics, and Isoplexis, which each include an equity interest. He receives research support from and consulted for Celgene and Roche, has received research support from Prelude Therapeutics, and has consulted for Incyte, Novartis, and Janssen. He has received honoraria from Lilly and Amgen for invited lectures and from Gilead for grant reviews. The remaining authors declare no competing financial interests.
The current affiliation for B.M.G. is Liverpool Hospital, Sydney, NSW, Australia.
The current affiliation for A.H. is Lund Stem Cell Center, Lund University, Lund, Sweden.
Correspondence: Wenbin Xiao, Hematopathology Diagnostic Service, Department of Pathology, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: xiaow@mskcc.org; and Mikhail Roshal, Hematopathology Diagnostic Service, Department of Pathology, Memorial Sloan Kettering Cancer Center, 1275 York Ave, New York, NY 10065; e-mail: roshalm@mskcc.org.