

Key Points
The detection of unrecognized inherited disorders in severe aplastic anemia calls for genetic testing in the diagnosis of all patients.
Patients with IBMFS, but not carriers, may need disease-specific transplant regimens to improve their outcomes.

Abstract
Patients with severe aplastic anemia (SAA) can have an unrecognized inherited bone marrow failure syndrome (IBMFS) because of phenotypic heterogeneity. We curated germline genetic variants in 104 IBMFS-associated genes from exome sequencing performed on 732 patients who underwent hematopoietic cell transplant (HCT) between 1989 and 2015 for acquired SAA. Patients with pathogenic or likely pathogenic (P/LP) variants fitting known disease zygosity patterns were deemed unrecognized IBMFS. Carriers were defined as patients with a single P/LP variant in an autosomal recessive gene or females with an X-linked recessive P/LP variant. Cox proportional hazard models were used for survival analysis with follow-up until 2017. We identified 113 P/LP single-nucleotide variants or small insertions/deletions and 10 copy number variants across 42 genes in 121 patients. Ninety-one patients had 105 in silico predicted deleterious variants of uncertain significance (dVUS). Forty-eight patients (6.6%) had an unrecognized IBMFS (33% adults), and 73 (10%) were carriers. No survival difference between dVUS and acquired SAA was noted. Compared with acquired SAA (no P/LP variants), patients with unrecognized IBMFS, but not carriers, had worse survival after HCT (IBMFS hazard ratio [HR], 2.13; 95% confidence interval[CI], 1.40-3.24; P = .0004; carriers HR, 0.96; 95% CI, 0.62-1.50; P = .86). Results were similar in analyses restricted to patients receiving reduced-intensity conditioning (n = 448; HR IBMFS = 2.39; P = .01). The excess mortality risk in unrecognized IBMFS attributed to death from organ failure (HR = 4.88; P < .0001). Genetic testing should be part of the diagnostic evaluation for all patients with SAA to tailor therapeutic regimens. Carriers of a pathogenic variant in an IBMFS gene can follow HCT regimens for acquired SAA.
Introduction
Severe aplastic anemia (SAA) is primarily an acquired immune-mediated bone marrow (BM) failure disorder with approximately 2 to 3 cases per million.1 The immunologic mechanisms contributing to SAA are not fully understood but are thought to be mediated through a combination of immune dysregulation, which ultimately leads to autoreactive cytotoxic T cell destruction of hematopoietic stem and progenitor cells (HSPC). Th1 and Th2 cells are increased while there is a reduction in the number and function of T regulatory cells, which normally suppress autoreactive T cells. Activated T cells in SAA also secrete myelosuppressive cytokines, and it has been hypothesized that there are changes in the BM microenvironment that influence the development of SAA.2,3 Increased risk of acquired SAA has been associated with exposure to certain medications and chemicals; however, this only contributes to a very small percentage of cases.1 Common genetic variants in the human leukocyte antigen (HLA) locus can confer an increased risk of developing SAA. HLA-A*02 and HLA-DRB1*0407, *15, and *1501 as well as HLA-DPB1 *03:01, *01:01, and HLA-B (rs28367832G>A) have been identified as SAA risk factors.4,5 Somatic loss of the HLA region on chromosome 6, occurring through copy neutral loss of heterozygosity (6pCNLOH), has been shown to be specific to acquired SAA and occurs in 8% to 15% of patients.4,6-8 In addition, a paroxysmal nocturnal hemoglobinuria (PNH) clone is also sensitive for identifying acquired over inherited disease.8-10 PNH is readily available clinically for patient identification, and currently, some clinical laboratories offer identification of mosaic 6pCNLOH.
Patients with inherited BM failure syndromes (IBMFS) develop aplastic anemia due to HSPC loss and/or destruction caused by inherited defects in key biological processes.11 The most common IBMFS are Fanconi anemia (a DNA repair disorder), dyskeratosis congenita (a telomere biology disorder), and the ribosome biology disorders Diamond Blackfan anemia and Shwachman Diamond syndrome. Many IBMFS often have associated congenital abnormalities and are often considered diseases of childhood. However, there is growing recognition that IBMFS can present without typical physical findings and/or family history, leading to delayed or misdiagnosis, particularly in adults.12 The phenotypic spectrum is often complicated by incomplete penetrance, variable expressivity, genetic anticipation, and/or overlapping phenotypes between IBMFS, primary immunodeficiencies, and inherited thrombocytopenias.13
Hematopoietic cell transplant (HCT) is the recommended first-line therapy for patients <40 years of age with acquired SAA and a matched sibling donor. For older patients with acquired disease or those without a suitable related donor, HCT is the second-line therapy, as it is for those who do not respond to immunosuppressive therapy or relapse.14 The 3-year survival probabilities for acquired SAA HCTs performed in the United States between 2013 and 2015 are 90% for matched sibling HCTs and 76% for unrelated donors.15 Recent work by Petit and colleagues and the Working Party of European Bone Marrow Transplantation showed 90% survival in a group of 74 patients transplanted with unrelated donors upfront.16 HCT is also an important therapeutic modality for patients with IBMFS who develop severe BM failure and is the only therapy that can mitigate the very high risk of myelodysplastic syndrome and acute myeloid leukemia in these patients. HCT regimens for patients with an IBMFS must be tailored to their underlying germline pathophysiology. Outcomes after HCT for individuals with IBMFS are improving, but the results, in most cases, are still worse than in patients with immune-mediated SAA.17-22
This study included 732 patients who underwent HCT for the indication of acquired SAA with an available blood sample stored at CIBMTR (Center for International Blood and Marrow Transplant Research) biorepository. We identified patients with unrecognized IBMFS and carriers of pathogenic or likely pathogenic (P/LP) variants in IBMFS genes and evaluated the effect of variant status on HCT outcomes.
Methods
SAA cohort
The TOAA (Transplant Outcome in Aplastic Anemia) study is a collaborative project between the NCI (National Cancer Institute) and CIBMTR that seeks to identify molecular predictors of HCT outcomes in patients with SAA.23 Clinical data and pre-HCT blood samples were provided by the CIBMTR (a research collaboration between the NMDP [National Marrow Donor Program; Be The Match] and the Medical College of Wisconsin). The CIBMTR actively audits all participating centers to ensure high data quality; details are available at https://www.cibmtr.org/DataManagement/AuditProgram/Pages/index.asp. The NMDP has collected biospecimens for all unrelated donor HCT performed in the United States since 1987 and from related donor HCT in the past decade. The current study included 732 patients with acquired SAA who received an unrelated (n = 636) or matched related (n = 96) donor peripheral blood stem cell or BM transplant with high-resolution HLA typing between 1989 and 2015 and who had pre-HCT DNA available for exome sequencing. All patients were diagnosed with acquired SAA based on standard clinical practices at the participating center and were not reported to have transformation to myelodysplastic syndrome or acute myeloid leukemia at HCT. Patients with known inherited disorders were excluded from the study, including those identified by the treating institution as having Fanconi anemia, dyskeratosis congenita, Diamond Blackfan anemia, or Shwachman Diamond syndrome. Family history, physical examination, and BM aspiration findings were not available for the study. Laboratory personnel was blinded to all patient characteristics and outcomes. All patients and donors provided informed consent for research participation, and the study was approved by the NMDP Institutional Review Board.
Sequencing
Exome sequencing data on all 732 patients were generated at the NCI Cancer Genomics Research Laboratory as previously described.24 In brief, DNA was extracted from pre-HCT peripheral blood mononuclear cells or whole blood using the QIAamp Maxi Kit (Qiagen Inc.; Valencia, CA). Exon-enriched libraries were generated via NimbleGen v3 or v3+UTR capture kit. Samples were sequenced (Illumina MiSeq or HiSeq) to an average depth of ∼55× and minimum coverage of >80% at 15×. Reads were aligned to the hg19 reference genome (Novoalign, Picard). GATK Version 3.8 UnifiedGenotyper, HaplotypeCaller, and Freebayes were used to call variants, with variants only used if called by at least 2 of 3 callers. Variant calling was limited to only the regions in the intersection of both the v3 and v3+UTR capture kit. Sanger sequencing was performed in a subset of related donor samples of SAA patients with a newly identified IBMFS variant to confirm genotype status.
All potentially deleterious variants were reviewed using IGV (Integrative Genomics Viewer; http://software.broadinstitute.org/software/igv/), and 34% of the variants had second orthogonal validation by AmpliSeq-targeted panel sequencing as previously described25; 99% of these were confirmed. All variants reported were present at an allelic balance consistent with germline origin with a variant allele frequency of 40% to 60% and had sufficient coverage. Variants with lower quality on IGV were sent for secondary validation. Exome sequencing of 597 in-house cancer-free samples (American Cancer Society’s CPS-II [Cancer Prevention Study-II] and the NCI PLCO [Prostate, Lung, Colorectal, and Ovarian Cancer] study) was used as a control to avoid the inclusion of false positives due to potential sequencing artifacts.26,27
Copy number variant (CNV) detection
VarSeq 2.2.1 (Golden Helix, Inc. [www.goldenhelix.com]; Bozeman, MT) was used to detect CNVs following the software instructions.28 For CNV detection, we limited the data to the 104 IBMFS genes using a browser extensible data file, including exonic regions of the genes of interest, generated by the University of California Santa Cruz genome browser (http://genome.ucsc.edu). After retrieving the depth of coverage data from BAM files and normalizing data using a reference set, we prioritized CNVs that: (1) were deletions, (2) passed software quality measures set by the software with no warning flags (high control variation, low control depth, low Z-score, and within the regional interquartile range [not significantly different from surrounding normal regions based on the regional interquartile range]), and (3) had P value < .001 (P value: probability that Z-scores would occur by chance in a diploid region). To identify putatively deleterious rare CNVs, filtered-in variants were individually reviewed according to the CNV annotation data based on the following databases: (1) gnomAD High Frequency CNV Regions 2019-11-25, GHI; (2) 1kG Phase3 CNVs and Large Variants 5b V2, GHI; (3) DECIPHER Population CNV v9.2; (4) RefSeq Genes 105.20201022 v2, NCBI; (5) ClinVar CNVs and Large Variants 2021-02-04, NCBI; (6) ClinGen Gene Dosage Sensitivity 2021-02-01, NCBI; or (7) Human Phenotype Ontology 2018-3-08. We used the CNV PhoRank algorithm to rank CNVs based on their relevance to IBMFS phenotypes as defined by the Human Phenotype Ontology. CNV PhoRank reports the ranking genes overlapping a given CNV, along with the sum and maximum score over all overlapping genes. To minimize the possibility of false positives, we included CNVs with a sum score >0.8 and removed those shared by several samples using the copy number probability/segregation algorithm.
Variant curation
We focused on 104 genes known to be associated with IBMFS across 4 major disease pathways: DNA damage response, hematopoiesis, ribosome biology, and telomere biology. These genes have 4 different modes of inheritance: 46 autosomal recessive (AR), 51 autosomal dominant (AD), 4 X-linked recessive (XLR), and 3 genes with both AD and AR inheritance (supplemental Tables 1 and 2, available on the Blood web site).
Variants were classified according to the ACMG (American College of Medical Genetics and Genomics) and the AMP (Association for Molecular Pathology) guidelines with a review of databases and literature for each variant.29 All variants were then manually reviewed, and the specific ACMG/AMP rules were adjusted based on the known disease and gene characteristics to reach a final interpretation.29 The rules that required patient-specific information or data of variant segregation within a family (eg, PS2, PM3, PM6, PP1, BS4, and BP2) could not be implemented because of the lack of information on the patients’ family history (supplemental Table 2A-B).
Variants were considered putatively causal based on pathogenicity reports in HGMD (Human Gene Mutation Database Professional 2021.1; http://www.hgmd.cf.ac.uk/ac/index.php), P/LP classification by ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/; last accessed March 1, 2021), loss-of-function (LOF) (frameshift, splice acceptor/donor site, or stop gain) in genes in which LOF is a known disease mechanism, location in a mutational hotspot and/or a critical and well-established functional domain, or different nucleotide change resulting in the same pathogenic amino acid change. All LOF variants qualified for the PVS1 rule.30 We used 5 in silico metapredictors (BayesDel, REVEL, CADD, MetaSVM, and EIGEN) for analysis.31-35 Missense variants that did not meet the ACMG/AMP P/LP criteria and had a deleterious in silico prediction in ≥3 out of 5 predictors were considered deleterious variants with unknown clinical significance (dVUS) (supplemental Table 2A).
Telomere length measurement
Telomere length was measured in the same DNA samples by monoplex quantitative polymerase chain reaction assay after adapting published methods as previously described.36 In brief, relative telomere length was calculated as the ratio of telomere signal concentration to that of a single copy gene (RPLP0); raw measurements were then standardized using internal quality‐controlled calibrator samples replicated within each plate. All telomeric and RPLP0 results were measured in triplicate, and their average was used for all calculations. Final relative telomere length values were log-transformed to ensure normality. Calculation of telomere length percentile-for-age was completed as described.37
Patient classification based on variant annotation and mode of inheritance
Patients were categorized into 3 groups according to variant status based on the known mode of inheritance for variants in each gene: (1) Unrecognized IBMFS: for example, a patient with 2 P/LP FANCA (AR gene) variants or a patient with a single P/LP GATA2 (AD gene) variant. For telomere biology genes with dual AD/AR inheritance, we also required relative telomere length to be <10th percentile-for-age to be considered an unrecognized IBMFS. (2) Carriers: individuals who had a single P/LP variant in an AR gene or females with a single P/LP variant in an XLR gene. (3) No variant: individuals with no P/LP variant(s) or those with dVUS. No variant individuals were presumed to have acquired immune-mediated SAA without an inherited cause of disease. Telomere length was not used in dVUS scoring. Survival analysis showed no difference between the dVUS group and the no variant group (supplemental Figure 1); hence these were combined into 1 group, which was then called “no variant” going forward.
Identification of somatic 6pCNLOH
Using genotyping data generated on the HumanOmniExpress-12v1-1_B array as described,4 we identified somatic 6pCNLOH using the MoChA software (https://github.com/freeseek/mocha) using a cell fraction >10% threshold.
Statistical analysis
The Kaplan-Meier estimator was used to calculate the probability of overall survival (OS) and 95% confidence intervals (CIs), with a log-rank test for statistical comparisons. Follow-up time started at the date of HCT and ended at death, censoring at the date of last follow-up or end of the study on 30 August 2017. In univariate analysis, we calculated 1, 3, and 5 years post-HCT probabilities of OS. Cox proportional hazard models were used for multivariable analyses, and all follow-up time was used. The selection of clinical factors included in the final model was based on a stepwise procedure with P = .25 for model entry and P = .15 for retention. The Schoenfeld residuals method was used to evaluate the proportional hazard assumption, and violation was addressed through stratification. The final model for OS was adjusted for recipient age, HLA matching, recipient race, conditioning intensity, donor age, Karnofsky Performance Score, recipient sex, and stratified on the year of transplant and donor type. For cause-specific mortality analyses, we modeled cause-specific hazards with causes of death other than those under study as competing events. For all models, the no variant group was set as the reference. Statistical analyses were performed using SAS 9.4 (SAS; Cary, NC) and R package “survminer”; P ≤ .01 was considered statistically significant to minimize multiple testing-related false discovery.
Data sharing
Deidentified sequencing data will be available through the dbGAP-controlled access database accession number dbGaP: phs001710.v2.p1.
Results
Pathogenic and likely pathogenic IBMFS gene variants in patients with SAA
Exome sequencing revealed 218 single nucleotide variants/small insertions/deletions (SNV/indels) and 10 CNVs in 59 IBMFS-associated genes (supplemental Tables 1 and 2). One hundred thirteen SNV/indels were P/LP per ACMG/AMP criteria (70% LOF) and were detected in 16.5% (121/732) patients. The remaining 105 variants were dVUS in 91 patients (12.4%).
Forty-eight (6.6%) patients met the criteria for an unrecognized IBMFS with 62 P/LP variants in 22 genes (Figure 1A). Twenty-one of 48 patients (44%) had P/LP variants in hematopoiesis genes, whereas 31% were in telomere biology genes (Figure 1B). In contrast, variants in carriers were more common in DNA damage response genes (42/73 [58%]) (Figure 1C). Nineteen unrecognized IBMFS patients had AR disease (16 compound heterozygous and 3 homozygous), 28 AD, and 1 XLR. Three patients had a P/LP SNV in compound heterozygosity with a large deletion in FANCA, SBDS, and ERCC6L2 (supplemental Table 3). Unrecognized IBMFS were most often because of P/LP variants in TINF2 (n = 6) and RUNX1 (n = 5), whereas variants in FANCM, SBDS, and ADA2 were most common in carriers (n = 10, 7, and 6, respectively) (Figure 1D-E and supplemental Table 1A-B).
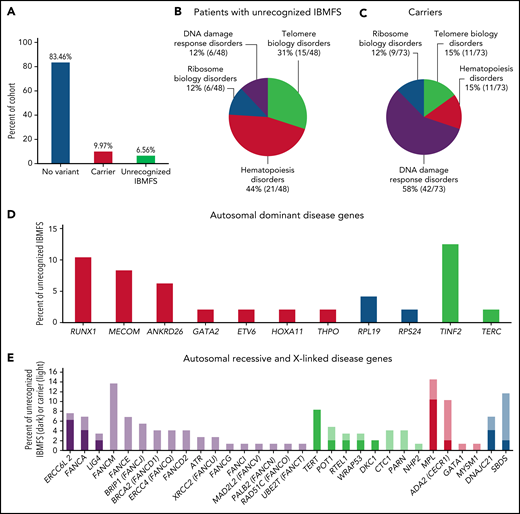
Frequencies and genes identified in the SAA cohort. (A) Frequency of variant status across the full cohort of patients with aplastic anemia. Blue, no variant; red, carrier (single pathogenic variant in an autosomal recessive gene or X-linked recessive gene in females); green, unrecognized IBMFS. (B) The frequency of SAA patients with unrecognized IBMFS and (C) carrier status categorized by gene pathway. (D) The number of patients with pathogenic/likely pathogenic variants in an autosomal dominant gene and (E) the number of patients with pathogenic/likely pathogenic variants in autosomal recessive, X-linked, or dual inheritance gene. Red, hematopoiesis genes; blue, ribosome biology genes; green, telomere biology genes; purple, DNA damage response gene. Darker bars, unrecognized IBMFS; lighter bars, carriers.
Frequencies and genes identified in the SAA cohort. (A) Frequency of variant status across the full cohort of patients with aplastic anemia. Blue, no variant; red, carrier (single pathogenic variant in an autosomal recessive gene or X-linked recessive gene in females); green, unrecognized IBMFS. (B) The frequency of SAA patients with unrecognized IBMFS and (C) carrier status categorized by gene pathway. (D) The number of patients with pathogenic/likely pathogenic variants in an autosomal dominant gene and (E) the number of patients with pathogenic/likely pathogenic variants in autosomal recessive, X-linked, or dual inheritance gene. Red, hematopoiesis genes; blue, ribosome biology genes; green, telomere biology genes; purple, DNA damage response gene. Darker bars, unrecognized IBMFS; lighter bars, carriers.
Patient characteristics and post-HCT survival by variant status
Seven hundred-thirty patients with available vital status contributed 3007 person-years of follow-up with 254 deaths. There were no statistically significant differences between age (P = .19), year of HCT (P = .21), or intensity of the HCT conditioning regimen (P = .83) in patients with an unrecognized IBMFS, carriers, or no variant (Tables 1 and 2 and Figure 2).
Demographics and transplant-related characteristics of SAA patients by IBMFS-associated variant status categories
| Variable | No Variant (n = 611) | Carrier (n = 73) | Unrecognized IBMFS (n = 48) | P* |
|---|---|---|---|---|
| Recipient age at transplant (yr), n (%) | .19 | |||
| ≤10 | 119 (19) | 15 (21) | 16 (33) | |
| >10, ≤20 | 181 (30) | 21 (29) | 16 (33) | |
| >20, ≤40 | 196 (32) | 23 (32) | 13 (27) | |
| >40 | 115 (19) | 14 (19) | 3 (6) | |
| Recipient sex, n (%) | .65 | |||
| Male | 340 (56) | 44 (60) | 25 (52) | |
| Female | 271 (44) | 29 (40) | 23 (48) | |
| Recipient race, n (%)† | .60 | |||
| Caucasian | 477 (78) | 60 (83) | 37 (77) | |
| Other | 131 (22) | 12 (17) | 11 (23) | |
| Karnofsky Performance Score, n (%)† | .81 | |||
| 10-80 | 126 (25) | 16 (28) | 8 (22) | |
| 90-100 | 369 (75) | 42 (72) | 29 (78) | |
| Donor type, n (%) | .94 | |||
| Related donor | 80 (13) | 9 (12) | 7 (15) | |
| Unrelated donor | 531 (87) | 64 (88) | 41 (85) | |
| Stem cell source, n (%)† | .20 | |||
| BM | 501 (82) | 66 (90) | 40 (83) | |
| Unknown | 109 (18) | 7 (10) | 8 (17) | |
| GvHD prophylaxis, n (%)† | .23‡ | |||
| Tacrolimus-based | 254 (42) | 37 (51) | 14 (29) | |
| CSA-based | 289 (47) | 32 (44) | 26 (54) | |
| Other | 61 (10) | 4 (5) | 8 (17) | |
| No GvHD prophylaxis | 6 (1) | 0 (0) | 0 (0) | |
| Conditioning regimen, n (%) | .85‡ | |||
| Myeloablative | 230 (38) | 29 (40) | 21 (44) | |
| RIC/nonmyeloablative | 377 (62) | 44 (60) | 27 (56) | |
| Other | 4 (1) | 0 (0) | 0 (0) | |
| Donor age at transplant (yr), n (%)† | .83 | |||
| ≤30 | 237 (42) | 30 (44) | 22 (50) | |
| >30, ≤40 | 164 (29) | 21 (31) | 10 (23) | |
| >40 | 158 (28) | 17 (25) | 12 (27) | |
| Donor sex, n (%)† | .56 | |||
| Male | 404 (67) | 51 (70) | 29 (60) | |
| Female | 202 (33) | 22 (30) | 19 (40) | |
| Donor/recipient CMV serostatus, n (%) | .20 | |||
| Donor negative/recipient negative | 167 (27) | 15 (21) | 16 (33) | |
| Donor negative/recipient positive | 181 (30) | 25 (34) | 12 (25) | |
| Donor positive/recipient negative | 72 (12) | 5 (7) | 8 (17) | |
| Donor positive/recipient positive | 149 (24) | 26 (36) | 9 (19) | |
| Unknown | 42 (7) | 2 (3) | 3 (6) | |
| Number of matches for HLA, n (%)†,§ | .31 | |||
| 8/8 | 335 (65) | 39 (63) | 22 (54) | |
| ≤7/8 | 177 (35) | 23 (37) | 19 (46) | |
| Year of transplant, n (%)† | .21 | |||
| 1989-2005 | 206 (34) | 27 (37) | 21 (44) | |
| 2006-2010 | 209 (34) | 17 (23) | 15 (31) | |
| 2011-2015 | 195 (32) | 29 (40) | 12 (25) |
| Variable | No Variant (n = 611) | Carrier (n = 73) | Unrecognized IBMFS (n = 48) | P* |
|---|---|---|---|---|
| Recipient age at transplant (yr), n (%) | .19 | |||
| ≤10 | 119 (19) | 15 (21) | 16 (33) | |
| >10, ≤20 | 181 (30) | 21 (29) | 16 (33) | |
| >20, ≤40 | 196 (32) | 23 (32) | 13 (27) | |
| >40 | 115 (19) | 14 (19) | 3 (6) | |
| Recipient sex, n (%) | .65 | |||
| Male | 340 (56) | 44 (60) | 25 (52) | |
| Female | 271 (44) | 29 (40) | 23 (48) | |
| Recipient race, n (%)† | .60 | |||
| Caucasian | 477 (78) | 60 (83) | 37 (77) | |
| Other | 131 (22) | 12 (17) | 11 (23) | |
| Karnofsky Performance Score, n (%)† | .81 | |||
| 10-80 | 126 (25) | 16 (28) | 8 (22) | |
| 90-100 | 369 (75) | 42 (72) | 29 (78) | |
| Donor type, n (%) | .94 | |||
| Related donor | 80 (13) | 9 (12) | 7 (15) | |
| Unrelated donor | 531 (87) | 64 (88) | 41 (85) | |
| Stem cell source, n (%)† | .20 | |||
| BM | 501 (82) | 66 (90) | 40 (83) | |
| Unknown | 109 (18) | 7 (10) | 8 (17) | |
| GvHD prophylaxis, n (%)† | .23‡ | |||
| Tacrolimus-based | 254 (42) | 37 (51) | 14 (29) | |
| CSA-based | 289 (47) | 32 (44) | 26 (54) | |
| Other | 61 (10) | 4 (5) | 8 (17) | |
| No GvHD prophylaxis | 6 (1) | 0 (0) | 0 (0) | |
| Conditioning regimen, n (%) | .85‡ | |||
| Myeloablative | 230 (38) | 29 (40) | 21 (44) | |
| RIC/nonmyeloablative | 377 (62) | 44 (60) | 27 (56) | |
| Other | 4 (1) | 0 (0) | 0 (0) | |
| Donor age at transplant (yr), n (%)† | .83 | |||
| ≤30 | 237 (42) | 30 (44) | 22 (50) | |
| >30, ≤40 | 164 (29) | 21 (31) | 10 (23) | |
| >40 | 158 (28) | 17 (25) | 12 (27) | |
| Donor sex, n (%)† | .56 | |||
| Male | 404 (67) | 51 (70) | 29 (60) | |
| Female | 202 (33) | 22 (30) | 19 (40) | |
| Donor/recipient CMV serostatus, n (%) | .20 | |||
| Donor negative/recipient negative | 167 (27) | 15 (21) | 16 (33) | |
| Donor negative/recipient positive | 181 (30) | 25 (34) | 12 (25) | |
| Donor positive/recipient negative | 72 (12) | 5 (7) | 8 (17) | |
| Donor positive/recipient positive | 149 (24) | 26 (36) | 9 (19) | |
| Unknown | 42 (7) | 2 (3) | 3 (6) | |
| Number of matches for HLA, n (%)†,§ | .31 | |||
| 8/8 | 335 (65) | 39 (63) | 22 (54) | |
| ≤7/8 | 177 (35) | 23 (37) | 19 (46) | |
| Year of transplant, n (%)† | .21 | |||
| 1989-2005 | 206 (34) | 27 (37) | 21 (44) | |
| 2006-2010 | 209 (34) | 17 (23) | 15 (31) | |
| 2011-2015 | 195 (32) | 29 (40) | 12 (25) |
BM, bone marrow; CMV, cytomegalovirus; CSA, cyclosporine; GvHD, graft-versus-host disease; RIC, reduced intensity conditioning; HLA, human leukocyte antigen.
Totals may not add up due to missing values.
χ-square test unless specified otherwise.
Kruskal-Wallis test.
Fisher’s exact test.
Follow-up and gene groupings of SAA patients by IBMFS-associated variant status categories
| Variable | No variant (n = 611) | Carrier (n = 73) | Unrecognized IBMFS (n = 48) | P |
|---|---|---|---|---|
| Alive at last follow-up, n (%)* | 403 (66) | 51 (70) | 22 (46) | .01† |
| Follow-up of survivors (mo), median (range) | 71 (2-295) | 58 (7-295) | 49 (9-125) | .06‡ |
| Gene group, n (%) | <.001§ | |||
| DNA damage response | 0 (0) | 42 (58) | 6 (13) | |
| Hematopoiesis | 0 (0) | 11 (15) | 21 (44) | |
| Ribosome biology | 0 (0) | 9 (12) | 6 (13) | |
| Telomere biology | 0 (0) | 11 (15) | 15 (31) | |
| No variant | 611 (100) | 0 (0) | 0 (0) |
| Variable | No variant (n = 611) | Carrier (n = 73) | Unrecognized IBMFS (n = 48) | P |
|---|---|---|---|---|
| Alive at last follow-up, n (%)* | 403 (66) | 51 (70) | 22 (46) | .01† |
| Follow-up of survivors (mo), median (range) | 71 (2-295) | 58 (7-295) | 49 (9-125) | .06‡ |
| Gene group, n (%) | <.001§ | |||
| DNA damage response | 0 (0) | 42 (58) | 6 (13) | |
| Hematopoiesis | 0 (0) | 11 (15) | 21 (44) | |
| Ribosome biology | 0 (0) | 9 (12) | 6 (13) | |
| Telomere biology | 0 (0) | 11 (15) | 15 (31) | |
| No variant | 611 (100) | 0 (0) | 0 (0) |
Totals may not add up due to missing values.
χ-square test unless specified otherwise.
Kruskal-Wallis test.
Fisher’s exact test.
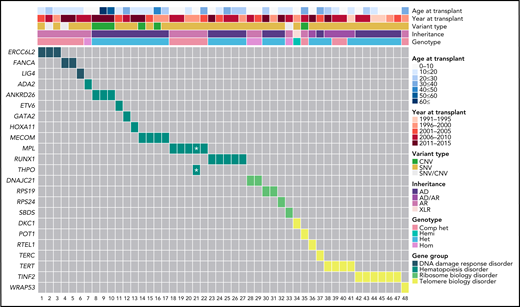
Genetic information by age and year of HCT of the 48 patients with SAA found to have an unrecognized IBMFS. Het, heterozygous; Comp het, compound heterozygous; Hemi, hemizygous; Hom, homozygous. *One patient had dual MPL and THPO inheritance. Note: HCT sample collection began in 1989, but the first patient in this group to have HCT was in 1991.
Genetic information by age and year of HCT of the 48 patients with SAA found to have an unrecognized IBMFS. Het, heterozygous; Comp het, compound heterozygous; Hemi, hemizygous; Hom, homozygous. *One patient had dual MPL and THPO inheritance. Note: HCT sample collection began in 1989, but the first patient in this group to have HCT was in 1991.
Patients with an unrecognized IBMFS had significantly worse post-HCT OS than those without an IBMFS (log-rank P = .0066) (Figure 3). Multivariable analysis confirmed this association (hazard ratio [HR], 2.13, 95% CI, 1.40-3.24; P = .0004) (Table 3). Reduced-intensity or nonmyeloablative conditioning regimens (RIC/NMC) did not mitigate the survival difference (HR, 2.05; 95% CI, 1.18-3.56; P = .01 in myeloablative condition [MAC]; and HR, 2.39; 95% CI, 1.21-4.69; P = .01 in RIC/NMC) (Table 3 and Figure 4). Carriers had no statistically significant post-HCT survival difference compared with patients with no variant, regardless of conditioning regimen (overall HR, 0.96; P = .86; MAC HR, 1.36; P = .27; RIC/NMC HR, 0.60; P = .24) (Figure 4 and Table 3). Similar results were observed in analyses restricted to patients receiving HCT from unrelated donors (HR, 2.20; P = .0003 unrecognized IBMFS; and HR, 1.01; P = .97 carriers). For the 96 patients receiving HCT from a matched related donor between 2008 and 2015 (samples from related donors were not available before 2008), all patients with unrecognized IBMFS (n = 7) survived during follow-up (supplemental Figure 2). Five of the 7 patients with unrecognized IBMFS had DNA available from their related donor; 1 donor was a carrier, and 4 donors did not have the variant identified in the recipient.

Overall survival by variant status. Kaplan-Meier curve comparing 3 groups of patients, those with a previously unrecognized IBMFS (green), carriers (red), and those with no variants (blue).
Overall survival by variant status. Kaplan-Meier curve comparing 3 groups of patients, those with a previously unrecognized IBMFS (green), carriers (red), and those with no variants (blue).
Multivariable survival analyses by IBMFS-associated variant status categories
| n Death/Total | All-cause Mortality | |||
|---|---|---|---|---|
| All patients | HR* (95% CI) | P | ||
| 26/48 | Unrecognized IBMFS | 2.13 (1.40-3.24) | .0004 | |
| 22/72 | Carrier | 0.96 (0.62-1.50) | .86 | |
| 205/606 | No variant | Reference | ||
| Unrelated donor | HR* (95% CI) | P | ||
| 26/41 | Unrecognized IBMFS | 2.20 (1.44-3.35) | .0003 | |
| 22/64 | Carrier | 1.01 (0.65-1.58) | .97 | |
| 197/527 | No variant | Reference | ||
| Myeloablative | HR† (95% CI) | P | ||
| 16/21 | Unrecognized IBMFS | 2.05 (1.18-3.56) | .01 | |
| 16/29 | Carrier | 1.36 (0.79-2.36) | .27 | |
| 108/228 | No variant | Reference | ||
| Reduced-intensity/nonmyeloablative | HR† (95% CI) | P | ||
| 10/27 | Unrecognized IBMFS | 2.39 (1.21-4.69) | .01 | |
| 6/43 | Carrier | 0.60 (0.26-1.40) | .24 | |
| 96/374 | No variant | Reference | ||
| DNA damage response | HR* (95% CI) | P | ||
| 4/6 | Unrecognized IBMFS | 4.43 (1.56-12.62) | .0053 | |
| 12/41 | Carrier | 0.84 (0.47-1.52) | .57 | |
| 205/606 | No variant | Reference | ||
| Hematopoiesis | HR* (95% CI) | P | ||
| 8/21 | Unrecognized IBMFS | 1.31 (0.64-2.70) | .46 | |
| 2/11 | Carrier | 0.59 (0.14-2.42) | .46 | |
| 205/606 | No variant | Reference | ||
| Ribosome biology | HR* (95% CI) | P | ||
| 3/6 | Unrecognized IBMFS | 2.22 (0.69-7.12) | .18 | |
| 3/9 | Carrier | 1.17 (0.37-3.76) | .79 | |
| 205/606 | No variant | Reference | ||
| Telomere biology | HR* (95% CI) | P | ||
| 11/15 | Unrecognized IBMFS | 2.84 (1.49-5.42) | .0015 | |
| 5/11 | Carrier | 1.87 (0.76-4.60) | .18 | |
| 205/606 | No variant | Reference | ||
| n Death/Total | All-cause Mortality | |||
|---|---|---|---|---|
| All patients | HR* (95% CI) | P | ||
| 26/48 | Unrecognized IBMFS | 2.13 (1.40-3.24) | .0004 | |
| 22/72 | Carrier | 0.96 (0.62-1.50) | .86 | |
| 205/606 | No variant | Reference | ||
| Unrelated donor | HR* (95% CI) | P | ||
| 26/41 | Unrecognized IBMFS | 2.20 (1.44-3.35) | .0003 | |
| 22/64 | Carrier | 1.01 (0.65-1.58) | .97 | |
| 197/527 | No variant | Reference | ||
| Myeloablative | HR† (95% CI) | P | ||
| 16/21 | Unrecognized IBMFS | 2.05 (1.18-3.56) | .01 | |
| 16/29 | Carrier | 1.36 (0.79-2.36) | .27 | |
| 108/228 | No variant | Reference | ||
| Reduced-intensity/nonmyeloablative | HR† (95% CI) | P | ||
| 10/27 | Unrecognized IBMFS | 2.39 (1.21-4.69) | .01 | |
| 6/43 | Carrier | 0.60 (0.26-1.40) | .24 | |
| 96/374 | No variant | Reference | ||
| DNA damage response | HR* (95% CI) | P | ||
| 4/6 | Unrecognized IBMFS | 4.43 (1.56-12.62) | .0053 | |
| 12/41 | Carrier | 0.84 (0.47-1.52) | .57 | |
| 205/606 | No variant | Reference | ||
| Hematopoiesis | HR* (95% CI) | P | ||
| 8/21 | Unrecognized IBMFS | 1.31 (0.64-2.70) | .46 | |
| 2/11 | Carrier | 0.59 (0.14-2.42) | .46 | |
| 205/606 | No variant | Reference | ||
| Ribosome biology | HR* (95% CI) | P | ||
| 3/6 | Unrecognized IBMFS | 2.22 (0.69-7.12) | .18 | |
| 3/9 | Carrier | 1.17 (0.37-3.76) | .79 | |
| 205/606 | No variant | Reference | ||
| Telomere biology | HR* (95% CI) | P | ||
| 11/15 | Unrecognized IBMFS | 2.84 (1.49-5.42) | .0015 | |
| 5/11 | Carrier | 1.87 (0.76-4.60) | .18 | |
| 205/606 | No variant | Reference | ||
Models were adjusted for recipient age, HLA matching, recipient race, conditioning intensity, donor age, Karnofsky Performance Score, and recipient sex and stratified on the year of transplant and donor type.
Models were adjusted for recipient age, recipient race, donor age, Karnofsky Performance Score, conditioning regimen, and recipient sex and stratified on the year of transplant and donor type.
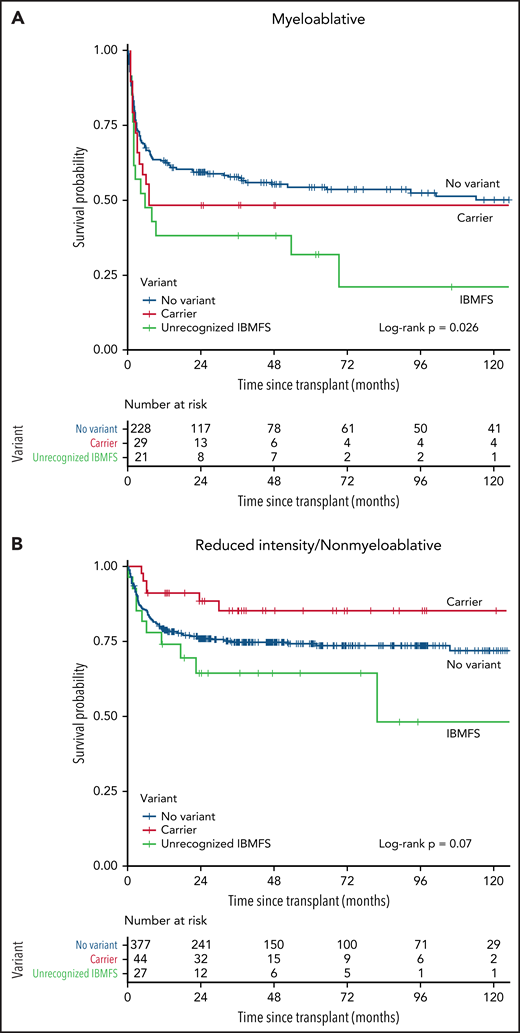
Survival probability after allogeneic HCT by IBMFS variant status and conditioning intensity. (A) Myeloablative and (B) reduced-intensity conditioning and nonmyeloablative Kaplan-Meier curve comparing 3 groups of patients, those with a previously unrecognized IBMFS (green), carriers (red), and those with no variants (blue).
Survival probability after allogeneic HCT by IBMFS variant status and conditioning intensity. (A) Myeloablative and (B) reduced-intensity conditioning and nonmyeloablative Kaplan-Meier curve comparing 3 groups of patients, those with a previously unrecognized IBMFS (green), carriers (red), and those with no variants (blue).
Analyses stratified by biologic pathway showed statistically significant inferior survival for those with an unrecognized IBMFS because of P/LP variant(s) in DNA damage response (HR, 4.43; 95% CI, 1.56-12.6; P = .0053) or telomere biology genes (HR, 2.84; 95% CI, 1.49-5.42; P = .0015), but not in carriers, regardless of the pathway (Table 3, supplemental Table 5, and Figure 5A,D). Patients with an unrecognized ribosome biology disease had an HR of 2.22 (P = .18), while those with variants in genes from hematopoiesis genes had an HR of 1.31 (P = .46) (Table 3, supplemental Table 4, and Figure 5B-C).
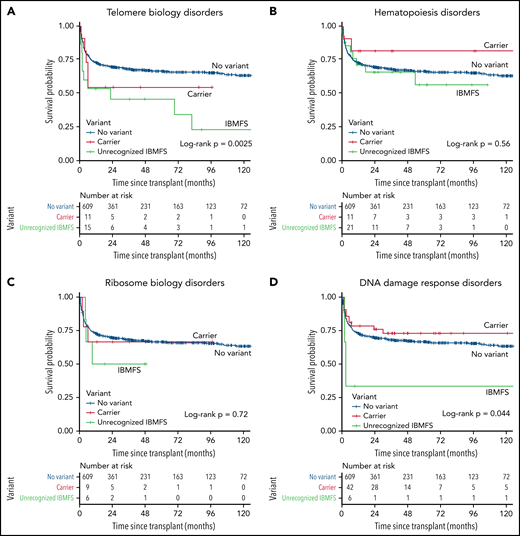
Posttransplant survival probability in patients with unrecognized IBMFS by gene group. (A) Telomere biology disorders. (B) Hematopoiesis disorders. (C) Ribosome biology disorders. (D) DNA damage response disorders. Kaplan-Meier curve comparing 3 groups of patients, those with a previously unrecognized IBMFS (green), carriers (red), and those with no variants (blue).
Posttransplant survival probability in patients with unrecognized IBMFS by gene group. (A) Telomere biology disorders. (B) Hematopoiesis disorders. (C) Ribosome biology disorders. (D) DNA damage response disorders. Kaplan-Meier curve comparing 3 groups of patients, those with a previously unrecognized IBMFS (green), carriers (red), and those with no variants (blue).
Cause of death analyses showed a statistically significant excess risk of death from organ failure in patients with unrecognized IBMFS (HR, 4.88; 95% CI, 2.20-10.82; P < .0001) but not in carriers (HR, 1.92; 95% CI, 0.73-5.03; P = .18), compared with patients with no variant. No statistically significant associations were noted for other causes of death by variant status (supplemental Table 5). Organ failure was the most common cause of death among patients with unrecognized IBMFS. In the no variant group, graft-versus-host disease (GvHD) followed by infection were the most common reasons for death (supplemental Figure 3). There was no significant difference in risk of death seen when comparing across the unrecognized IBMFS by gene groups (supplemental Table 6). Multivariate analysis showed no associations between IBMFS variant status and neutrophil or platelet engraftment or acute or chronic GvHD.
Fifty-six of 732 (7.6%) patients had 6pCNLOH with a cell fraction >10%. Within the acquired SAA subgroup (carriers plus no variant), 8.2% (56/684) had 6pCNLOH, consisting of 5.5% (4/73) carriers with 6pCNLOH, and 8.5% (52/611) of the no variant group (supplemental Figure 4). PNH data were not available for the cohort.
Discussion
In this real-world cohort of HCT recipients for clinically diagnosed SAA, nearly 7% of the patients had an undiagnosed germline genetic cause of their BM failure that negatively affected their outcome, and approximately one-third were adults at HCT. These findings quantify the importance of germline genetic testing for all individuals with SAA as part of their diagnostic evaluation and before HCT. Patients should have clinical diagnostic testing for specific diseases when available, including chromosome breakage analysis, telomere length testing, erythrocyte ADA, and exocrine pancreatic insufficiency testing. Patients with PNH clones may not need germline genetic testing, as the presence of these clones is extremely rare in IBMFS.9 Our findings have direct clinical implications because patients with IBMFS do not typically respond to immunosuppressive therapy (IST), require modified or disease-specific conditioning regimens, and are at risk of disease-specific complications (eg, solid malignancies in Fanconi anemia or pulmonary fibrosis in telomere biology disorders).38,39 Treatment records before HCT were not available in our registry-based study. Therefore, we assumed that most patients in this study had not responded to IST because IST is the first-line standard of care for patients with SAA and no matched related donor.1 Patients with SAA because of IBMFS do not respond to IST, further illustrating the importance of early genetic diagnosis to guide therapeutic decisions.
Our finding of nearly 7% of SAA patients with unrecognized IBMFS is remarkably consistent with the frequency of undiagnosed cancer predisposition syndromes identified in patients with pediatric cancers.40 The understanding of the prevalence of cancer predisposition syndromes in children and adults has increased germline genetic testing in those patients, and our data suggest a need for this in SAA.
Differentiating acquired SAA from an IBMFS is often complicated by the lack of physical findings and nongenetic laboratory tests, particularly in disorders of hematopoiesis (eg, GATA2 or RUNX1). Variable disease penetrance and expressivity further reduce the ability to identify patients through family history. Additionally, BM failure often precedes the development of other IBMFS clinical features.41 The inferior survival of patients with an unrecognized IBMFS was primarily driven by the relatively high frequency of variants in DNA damage repair genes and telomere biology disorders, which is consistent with the previously reported poor post-HCT survival in patients with dyskeratosis congenita.17 Most patients with unrecognized IBMFS in this study were not patients with Fanconi anemia underscoring the effectiveness of chromosome breakage testing in detecting most of those patients upfront. Chromosome breakage testing does not identify all DNA damage response disorders (eg, ERCC6L2 or LIG4). This study showed a significant association between death from organ failure and having an unrecognized IBMFS, which may be related to their underlying pathophysiology.
We acknowledge that some of the patients in our study underwent HCT before the first description of their disease-associated gene and could not have been identified by genetic testing at diagnosis. However, our findings showed that the observed survival difference by IBMFS was independent of the year of transplant. We did identify patients who had HCT after the discovery of their disease gene who could have benefitted from disease-specific regimens and post-HCT surveillance. Interestingly, we identified 3 unrecognized IBMFS cases because of compound heterozygous SNV/CNVs. This underscores the importance of including CNV analysis in genetic testing. Our data also show that the 105 variants scored as deleterious by in silico criteria only (dVUS) were not associated with differences in post-HCT survival, further supporting the use of ACMG/AMP variant curation guidelines.29
Of note, while nonmyeloablative regimens are now standard for SAA, 40% and 38% of patients with unrecognized IBMFS and acquired SAA, respectively, received myeloablative regimens in this study. While it is not possible to ascertain the rationale for choosing a higher-intensity regimen based on the available data, this is not expected to affect our results because our data showed the use of RIC/NMC regimens did not close the survival gap between unrecognized IBMFS and acquired SAA patients (ie, no variant). It is important to recognize that patients with an IBMFS have improved outcomes with the use of disease-specific HCT regimens, and the unrecognized IBMFS patients in this cohort would have benefitted from such tailored regimens.42
Our data do not show a statistically significant survival difference in carriers compared with those with no variant. This answers the longstanding important clinical question of whether patients with SAA carrying a single P/LP variant in an autosomal recessive gene or females with X-linked variants have worse outcomes than patients without these variants. A future study is required to answer the reverse question: does having an HCT donor who is a germline carrier affect recipient post-HCT outcome? Sibling transplants for many autosomal recessive diseases, such as Fanconi anemia, have been done for decades, but data on whether heterozygous P/LP variant status is associated with recipient outcomes have not been quantified.42 Additionally, it will be interesting to see if somatic hemopoietic mosaicism in the donor affects HCT outcomes in SAA.
The strengths of our study include the large sample size in a rare disease, comprehensive gene and variant assessment with a conservative ACMG/AMP approach, and detailed clinical outcome data. A limitation of our study is the use of only blood samples for germline exome sequencing; however, none of the variants identified are typically seen in clonal hematopoiesis of indeterminate potential, and particular care was taken to assure that bioinformatic pipelines were optimized for germline variants.43 We were not able to measure telomere length with the clinically approved flow cytometry with florescent in situ hybridization method because of a lack of fresh or cryopreserved blood samples. This may have led to the misclassification of some of the patients with unrecognized telomere biology disorder as carriers. Our conservative approach to variant interpretation may have led to an undercounting of potentially significant variants. Future comprehensive functional studies on all dVUS will be necessary to completely understand whether they have a role in disease etiology or outcomes. As physical exam or clinical diagnostic testing data, such as chromosome breakages testing, was not available to us, we relied on the transplant center for assignment of acquired SAA as the reason for HCT. It is possible that some patients with the known inherited disease were included, which would inflate the unrecognized IBMFS identified by sequencing. However, the data collection mechanism used by CIBMTR is distinct for those with inherited vs acquired SAA. We used causes of death information reported by the transplant center that may be limited in accuracy; however, a recent study with an adjudication committee showed an 87.5% agreement overall with reported causes of death.44
Conclusions
This study conclusively shows the importance of comprehensive germline genetic testing for all patients with SAA regardless of age, physical phenotype, comorbid conditions, or family history, except for those with PNH or 6pCNLOH clones. HCT for patients with SAA should be informed by genetic testing as these data show inferior outcomes for those with unrecognized inherited disease compared with immune-mediated SAA.
Acknowledgments
The authors thank the study participants. This work used the computational resources of the National Institutes of Health (NIH) High Performance Computing Biowulf cluster. Investigators from the CPS-II (Cancer Prevention Study-II) and the cohort express sincere appreciation to all participants and to each member of the study and biospecimen management group.
This work was supported by the Intramural Research Program of the NCI (National Cancer Institute). The NCI Prostate, Lung, Colorectal, and Ovarian Cancer study is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and by contracts from the Division of Cancer Prevention, NCI, NIH. The Cancer Genomics Research Laboratory is funded with federal funds from the NCI, NIH under NCI Contract 75N910D00024. The Center for International Blood and Marrow Transplant Research is supported primarily by Public Health Service U24CA076518 from the NCI, the National Heart, Lung, and Blood Institute, and the National Institute of Allergy and Infectious Diseases, HHSH250201700006C from the Health Resources and Services Administration, and N00014-20-1-2705 and N00014-20-1-2832 from the Office of Naval Research. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort.
Authorship
Contribution: L.J.M.R., M.R., and Y.W. analyzed the data, made the figures and tables, and wrote the manuscript; S.M.G. and S.A.S. supervised the project; B.J.B., V.V.W., and J.K. aided with variant identification; W.Z. and R.M.H. performed and analyzed the mosaic chromosomal event identification; B.H., B.Z., and K.J. managed the DNA sequencing and bioinformatics; C.D. performed telomere length testing; B.C., S.S., and N.D.F. provided in-house control samples; T.W., S.P., M.H., S.G.E.M., S.R.S., and S.J.L. facilitated the collaboration and transfer of samples and clinical data; and all authors reviewed, edited, and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Lisa J. McReynolds, Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, 9609 Medical Center Drive, 6E434, Bethesda, MD 20892; e-mail: lisa.mcreynolds@nih.gov.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal