

Key Points
Two-year fixed-duration VenR offers sustained PFS, OS, and MRD benefits over BR in R/R CLL, regardless of presence of high-risk biomarkers.
Median MRD doubling time post-VenR EOT was 93 days, with 19 months from uMRD to MRD conversion and another 25 months from conversion to PD.

Abstract
The MURANO trial (A Study to Evaluate the Benefit of Venetoclax Plus Rituximab Compared With Bendamustine Plus Rituximab in Participants With Relapsed or Refractory Chronic Lymphocytic Leukemia [CLL]; ClinicalTrials.gov identifier #NCT02005471) reported superior progression-free survival (PFS) and overall survival (OS) with venetoclax-rituximab (VenR) vs bendamustine-rituximab (BR) in relapsed/refractory (R/R) CLL. Patients were randomized to 2 years of VenR (n = 194; rituximab for the first 6 months) or 6 months of BR (n = 195). Although undetectable minimal residual disease (uMRD) was achieved more often with VenR, the long-term implications of uMRD with this fixed-duration, chemotherapy-free regimen have not been explored. We report MRD kinetics and updated outcomes with 5 years’ follow-up. Survival benefits with VenR vs BR were sustained (median PFS [95% confidence interval]: 53.6 [48.4, 57.0] vs 17.0 [15.5, 21.7] months, respectively, P < .0001; 5-year OS [95% confidence interval]: 82.1% [76.4, 87.8] vs 62.2% [54.8, 69.6], P < .0001). VenR was superior to BR, regardless of cytogenetic category. VenR-treated patients with uMRD at end of treatment (EOT; n = 83) had superior OS vs those with high-MRD+ (n = 12): 3-year post-EOT survival rates were 95.3% vs 72.9% (P = .039). In those with uMRD at EOT, median time to MRD conversion was 19.4 months. Of 47 patients with documented MRD conversion, 19 developed progressive disease (PD); median time from conversion to PD was 25.2 months. A population-based logistic growth model indicated slower MRD median doubling time post-EOT with VenR (93 days) vs BR (53 days; P = 1.2 × 10−7). No new safety signals were identified. Sustained survival, uMRD benefits, and durable responses support 2-year fixed-duration VenR treatment in R/R CLL.
Introduction
Recent treatment advances in relapsed or refractory (R/R) chronic lymphocytic leukemia (CLL) have led to improved outcomes and prolonged progression-free survival (PFS).1 Minimal residual disease (MRD) is often used in CLL clinical trials of time-limited therapies to quantify depth of treatment response,2 with MRD at the end of treatment (EOT) a predictor of long-term clinical outcomes.3-7 Achievement of undetectable MRD (uMRD) (<10−4)4 is an important goal of time-limited therapy, as it is associated with improved PFS and overall survival (OS).8 Serial MRD assessment identifies patients with increasing subclinical disease burden months before clinical manifestation of recurrence.9
Constitutively overexpressed in CLL, B-cell lymphoma 2 (BCL-2) is an anti-apoptotic protein and key regulator of the intrinsic apoptotic pathway.10,11 Unmutated (unmut) immunoglobulin heavy chain gene (IGHV), deletion in chromosome 17p [del(17p)], and genomic complexity (GC) are prognostic factors that can negatively influence response to treatment.12-18 GC, defined by the presence of ≥3 copy number alterations (CNA), is often an indication of aggressive CLL and is consistently linked with reduced time to next treatment (TTNT) and poorer survival outcomes.17,18 Although unmut-IGHV status has historically been an unfavorable marker in predicting survival outcomes after chemoimmunotherapy in CLL,19,20 the impact varies with treatment type, and novel agents targeting B-cell receptor pathway kinases have lessened its influence on response rates and PFS.13,21,22 Up to 80% of del(17p) cases also harbor a mutation in TP53, encoding the tumor suppressor protein p53, which plays a critical role in maintaining genomic integrity.23,24 Furthermore, TP53 mutations can occur without del(17p) and retain an adverse prognostic impact.24,25 Venetoclax (Ven) is a highly selective BCL-2 inhibitor that induces high response rates in patients with R/R CLL.13,26 Acting independently of TP53,27 Ven can induce high response rates in patients with R/R CLL carrying del(17p), although TP53 aberrations can still affect survival outcomes post-Ven treatment.26,28
The phase 3 MURANO trial (A Study to Evaluate the Benefit of Venetoclax Plus Rituximab Compared With Bendamustine Plus Rituximab in Participants With Relapsed or Refractory Chronic Lymphocytic Leukemia [CLL]) investigated the efficacy and safety of fixed-duration therapy with Ven plus rituximab (VenR) for 6 months followed by single-agent Ven for up to a total duration of 2 years, compared with 6 months of bendamustine plus rituximab (BR), in patients with R/R CLL.5,29,30 The MURANO primary analysis reported significantly longer PFS with VenR vs BR, with benefit observed in all subgroups analyzed, including patients with unmut-IGHV or del(17p)/TP53 mutation (TP53-mut).30 PFS and OS benefits of VenR were sustained at 3 and 4 years of follow-up.5,29 Moreover, uMRD was observed in peripheral blood (PB) at higher rates in the VenR arm than in the BR arm at the end of combination treatment (EOCT).29 Genetic risk factors involving TP53 and GC affected MRD rates and PFS in both treatment arms.5
To assess clinical relevance of markers of depth of remission (eg, MRD status) and durability of uMRD, as well as evaluate characteristics of patients with early relapse, longer follow-up of the MURANO cohort is required. Here, we report MRD kinetics and updated efficacy and safety outcomes over 5 years (with 3 years’ posttreatment), including subgroup analyses of patients with high-risk biomarkers.
Methods
Study design and conduct
The study design, eligibility criteria, dosing, prophylactic measures, and monitoring for MURANO (ClinicalTrials.gov identifier #NCT02005471) have been published previously.30 The trial was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation of Good Clinical Practice guidelines. The trial protocol was approved by the ethics committee at each participating institution, and written informed consent was provided by all patients. The clinical cutoff date for the current analysis was May 8, 2020.
End points and assessments
The primary efficacy end point was investigator-assessed PFS, defined as time from randomization to first occurrence of progressive disease (PD) or death.
PB MRD status was a secondary end point, assessed at cycle 4, 2 to 3 months after EOCT, and every 3 to 6 months thereafter (including EOT); it was analyzed centrally by allele-specific oligonucleotide polymerase chain reaction and/or flow cytometry.29,30 Patients were categorized according to MRD status as previously defined,4 where the uMRD cutoff was <10−4 and MRD+ was ≥10−4; low-MRD+ was 10−4 to <10−2, and high-MRD+ was ≥10−2. MRD conversion was defined as 2 consecutive assays detecting MRD at ≥10−4 or PD according to International Workshop on CLL (iwCLL) criteria31 in patients who previously had uMRD.
Other secondary end points included TTNT, OS, complete response (CR), and partial response rates (according to iwCLL 2008 criteria31 and assessed by computed tomography scan at screening, cycle 4, and at EOCT) and safety assessments.
Safety data collected for the current analysis period (posttreatment only, not including any adverse events [AEs] occurring during treatment) were prespecified AEs of concern, serious AEs related to the study drug, and development of a second primary malignancy. Further details of disease status assessment and safety monitoring are included in the supplemental Methods (available on the Blood Web site) or have been described previously.29,30
MRD doubling time analysis
For longitudinal analysis of MRD growth dynamics post-EOT, and to better understand the clinical differences noted in patient outcomes, a population-based logistic growth model with a nonlinear mixed effects approach was developed as previously described.32 Patients who completed treatment without prior PD and who had ≥2 measurable time points (assay lower limit of quantification of ∼10−5 for allele-specific oligonucleotide polymerase chain reaction) post-EOT were included; data below the lower limit of quantification were managed by using a likelihood-based imputation method.33 For analyses of covariates, disease burden was defined by tumor lysis syndrome risk definition.34,35 Further details are available in the supplemental Methods.
GC and molecular assessments
IGHV mutation status was assessed by PCR and TP53 status by next-generation sequencing. Molecular assessments, including analysis of GC and del(17p), were conducted by using high-density array comparative genomic hybridization; data processing was performed as previously published.5 GC was defined as ≥3 CNA and high GC as ≥5 CNA.17,18
Response to next-line therapy
Patients in either treatment arm who experienced PD were followed for response to any subsequent anti-CLL therapeutic regimens. In addition, in a substudy of the MURANO population, patients from the VenR arm could be retreated with the VenR regimen at the same dose and schedule as the main study, and patients in the BR arm could cross over to receive the VenR regimen in the case of progression with indication for treatment according to the iwCLL criteria.4 Patients who initiated a new anti-CLL therapy but who did not have a response assessment reported by the investigator were considered unevaluable.
Statistical analysis
There was no α spending allocated to the current analysis, and therefore all P values are considered descriptive. Kaplan-Meier estimates analyzed time-to-event data, including landmark analyses from EOCT and EOT according to MRD status. The log-rank test and Cox proportional hazards regression model compared overall PFS and OS across treatment arms. Fisher’s exact test was performed to compare MRD status at EOCT and EOT, and clinical and cytogenetic risk factors in VenR-treated patients with and without PD after EOT. Statistical inference on MRD doubling time was derived for treatment arms (and other stratified subgroups) by using the unpaired t-test.
Results
Patient characteristics and follow-up
In total, 389 patients were enrolled; 194 received VenR and 195 received BR (supplemental Figure 1). Overall, 130 patients completed 2 years of Ven treatment without PD. Median duration of follow-up from study enrollment for the current analysis was 59.2 months (range, 0 to 71.5 months).
PFS and TTNT
With patients off treatment for 3 years, the PFS benefit with VenR treatment over BR was sustained (hazard ratio [HR], 0.19; 95% confidence interval [CI], 0.15, 0.26; P < .0001) (Figure 1A). Median PFS was 53.6 months (95% CI, 48.4, 57.0) for VenR-treated patients and 17.0 months (95% CI, 15.5, 21.7) for BR-treated patients. Consistent PFS benefits with VenR were also observed in subgroup analyses according to patient demographics, biomarkers, and baseline characteristics (supplemental Figure 2). Estimated PFS at 3 years’ post-EOT (5 years from the start of treatment) in the 194 patients assigned to VenR was 37.8% (95% CI, 28.8, 46.8); among the 130 patients who completed the full 2 years of Ven treatment, this was 51.1% (95% CI, 40.2, 61.9). VenR treatment compared with BR treatment significantly improved TTNT (HR, 0.26; 95% CI, 0.20, 0.35; P < .0001). Median TTNT was 57.8 months (95% CI; 55.1, not estimable [NE]) for VenR-treated patients and 23.9 months (95% CI, 20.7, 29.5) for BR-treated patients (supplemental Figure 3).

Kaplan-Meier estimates of investigator-assessed PFS in the overall intention-to-treat population (A) and according to IGHV mutation status (B), del(17p) and/or TP53 mutation status (C), and GC status (D).P values are descriptive only. *Stratified analysis; unstratified HR, 0.21 (95% CI, 0.16, 0.27). †Unstratified analysis. +, censored.
Kaplan-Meier estimates of investigator-assessed PFS in the overall intention-to-treat population (A) and according to IGHV mutation status (B), del(17p) and/or TP53 mutation status (C), and GC status (D).P values are descriptive only. *Stratified analysis; unstratified HR, 0.21 (95% CI, 0.16, 0.27). †Unstratified analysis. +, censored.
Across both treatment arms, among 39 (13.5%) patients with del(17p), 31 (79.5%) also had GC, and 12 (30.8%) of 39 had high GC. Presence of high-risk cytogenetic and/or molecular abnormalities were associated with shorter median PFS among patients treated with VenR: unmut-IGHV vs mutated (mut), 52.2 months vs NE, respectively (HR [95% CI], 2.96 [1.64, 5.34], P = .0002); del(17p) and/or TP53-mut vs TP53-wild-type (WT), 37.4 vs 56.6 months (HR [95% CI], 2.04 [1.32, 3.15], P = .010); and GC vs no GC, 41.7 vs 59.8 months (HR [95% CI], 2.50 [4.00, 1.56]; P < .0001). Although similar patterns were observed with BR treatment, median PFS was consistently inferior for each subset compared with VenR (Figure 1B-D). Five-year PFS rates with VenR were lower in patients with these high-risk characteristics, ranging from 17.7% to 28.7% vs 42.5% to 72.7% for those without.
Overall survival
The OS benefit for patients treated with VenR vs BR was maintained (HR, 0.40; 95% CI, 0.26, 0.62; P < .0001), with 5-year OS estimates of 82.1% (95% CI, 76.4, 87.8) for VenR and 62.2% (95% CI, 54.8, 69.6) for BR (Figure 2A). Median OS was not reached for either treatment arm.
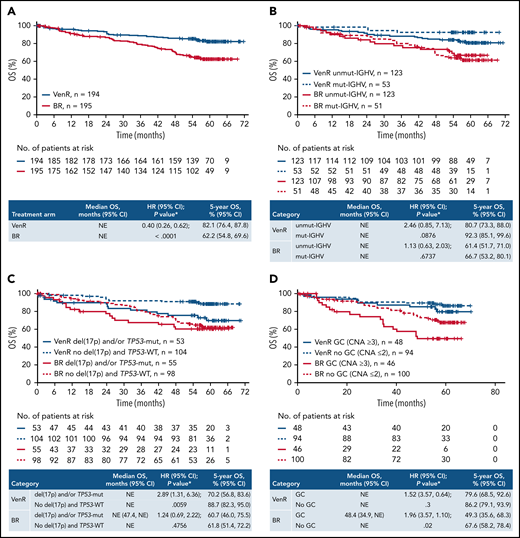
Kaplan-Meier estimates of OS in the overall intention-to-treat population (A) and according to IGHV mutation status (B), del(17p) and/or TP53 mutation status (C), and GC status (D).P values are descriptive only. *Stratified analysis; unstratified HR, 0.42 (95% CI 0.27, 0.64). †Unstratified analysis. +, censored.
Kaplan-Meier estimates of OS in the overall intention-to-treat population (A) and according to IGHV mutation status (B), del(17p) and/or TP53 mutation status (C), and GC status (D).P values are descriptive only. *Stratified analysis; unstratified HR, 0.42 (95% CI 0.27, 0.64). †Unstratified analysis. +, censored.
VenR also resulted in improved OS vs BR across various subgroups according to patient demographics, biomarkers, and baseline characteristics (supplemental Figure 4).
Assessment of OS according to IGHV mutation status, del(17p) and/or TP53-mut, and GC, showed a consistently more favorable outcome with VenR vs BR, with 5-year estimates of 70.2% to 92.3% with VenR compared with 49.3% to 67.6% with BR (Figure 2B-D). Among patients treated with VenR, those with mut-IGHV had the highest 5-year OS estimate (92.3%), and patients with del(17p) and/or TP53-mut had the lowest estimate (70.2%).
Landmark analysis of PFS and OS by PB MRD status at EOT
VenR-treated patients with uMRD at EOT (n = 83) had a 3-year post-EOT PFS estimate of 61.3% (95% CI, 47.3, 75.2) vs 40.7% (95% CI, 19.2, 62.2) in low-MRD+ patients (n = 23), whereas all but 1 patient with high-MRD+ (n = 12) had PD before 2 years’ post-EOT (Figure 3A). Particularly durable PFS was observed among patients with IGHV-mut who completed treatment and had uMRD at EOT (supplemental Figure 5A).

Kaplan-Meier estimates of landmark PFS (A) and OS (B) according to PB MRD level at EOT in patients who completed 2 years of Ven treatment without prior PD. *Stratified. +, censored.
Kaplan-Meier estimates of landmark PFS (A) and OS (B) according to PB MRD level at EOT in patients who completed 2 years of Ven treatment without prior PD. *Stratified. +, censored.
Among VenR-treated patients, OS was better for those with uMRD at EOT vs low-MRD+ or high-MRD+, with 3-year post-EOT estimates of 95.3% (95% CI, 90.0, 100.0), 91.3% (95% CI, 79.8, 100.0), and 72.9% (95% CI, 46.4, 99.5), respectively (Figure 3B). There was a statistically significant difference between the patients with uMRD vs high-MRD+.
MRD conversion after EOT and association with clinical outcomes
Baseline disease characteristics of VenR-treated patients according to MRD response status at EOT are provided in supplemental Table 1.
Eighty-three patients (42.8%) completed the protocol-specified 2 years of Ven treatment without prior PD and had uMRD at EOT. At the 5-year update, 4 (4.8%) of these patients had CLL PD without prior confirmed MRD conversion (none of whom had Richter’s transformation), 47 (56.6%) had confirmed MRD conversion, and 32 (38.6%) remained in uMRD status without PD. Median time from EOT to MRD conversion was 19.4 months (95% CI, 8.7, 28.3) (Figure 4A). Of the 47 patients with documented MRD conversion, 19 (40.4%) subsequently developed PD according to iwCLL criteria, with a median time from conversion to PD of 25.2 months (95% CI, 19.4, 30.4) (Figure 4B). MRD conversion kinetics in this patient subset are shown in supplemental Figure 6.

Kaplan-Meier estimates of time from EOT to MRD conversion (A) and time from MRD* conversion to PD† (B) in VenR-treated patients (n = 83) who had uMRD at EOT. *MRD conversion was defined as 2 consecutive assays detecting MRD at ≥10−4 or PD according to iwCLL criteria31 in patients who previously had uMRD. †Investigators assessed PD by using iwCLL criteria.31 uMRD, <1 CLL cell/10 000 leukocytes. +, censored.
Kaplan-Meier estimates of time from EOT to MRD conversion (A) and time from MRD* conversion to PD† (B) in VenR-treated patients (n = 83) who had uMRD at EOT. *MRD conversion was defined as 2 consecutive assays detecting MRD at ≥10−4 or PD according to iwCLL criteria31 in patients who previously had uMRD. †Investigators assessed PD by using iwCLL criteria.31 uMRD, <1 CLL cell/10 000 leukocytes. +, censored.
Among patients with uMRD at EOT, unmut-IGHV, del(17p), and GC at baseline were all associated with an increased likelihood of developing PD (Table 1). The rate of MRD conversion with eventual PD was higher in those with unmut-IGHV (21 of 56 [37.5%]) vs those with mut-IGHV (1 of 23 [4.3%]) (supplemental Figure 5B-C). The 4 patients with del(17p) who were uMRD at EOT all experienced MRD conversion with subsequent PD, whereas 8 (44.4%) of 18 patients with GC converted to MRD+ and developed PD, compared with 8 (20.0%) of 40 patients without GC.
MRD conversion and PD status according to patients’ genetic profile status at baseline (VenR arm)
| IGHV | GC (≥3 CNA) | del(17p) | |||||
|---|---|---|---|---|---|---|---|
| MRD status | ITT N = 194 | Unmut n = 123* | Mut n = 53* | GC n = 48* | No GC n = 94* | Present n = 17* | Absent n = 125* |
| uMRD at EOT | 83 (42.8%) | 56 (45.5%) | 23 (43.4%) | 18 (37.5%) | 40 (42.5%) | 4 (23.5%) | 54 (43.2%) |
| Sustained uMRD | 32 (16.5%) | 20 (35.7%) | 10 (43.5%) | 5 (27.8%) | 16 (40.0%) | 0 (0%) | 21 (38.9%) |
| Conversion to MRD (no PD) | 28 (14.4%) | 15 (26.8%) | 12 (52.2%) | 5 (27.8%) | 16 (40.0%) | 0 (0%) | 21 (38.9%) |
| Conversion with subsequent PD | 19 (9.8%) | 21 (37.5%) | 1 (4.3%) | 8 (44.4%) | 8 (20.0%) | 4 (100%) | 12 (22.2%) |
| IGHV | GC (≥3 CNA) | del(17p) | |||||
|---|---|---|---|---|---|---|---|
| MRD status | ITT N = 194 | Unmut n = 123* | Mut n = 53* | GC n = 48* | No GC n = 94* | Present n = 17* | Absent n = 125* |
| uMRD at EOT | 83 (42.8%) | 56 (45.5%) | 23 (43.4%) | 18 (37.5%) | 40 (42.5%) | 4 (23.5%) | 54 (43.2%) |
| Sustained uMRD | 32 (16.5%) | 20 (35.7%) | 10 (43.5%) | 5 (27.8%) | 16 (40.0%) | 0 (0%) | 21 (38.9%) |
| Conversion to MRD (no PD) | 28 (14.4%) | 15 (26.8%) | 12 (52.2%) | 5 (27.8%) | 16 (40.0%) | 0 (0%) | 21 (38.9%) |
| Conversion with subsequent PD | 19 (9.8%) | 21 (37.5%) | 1 (4.3%) | 8 (44.4%) | 8 (20.0%) | 4 (100%) | 12 (22.2%) |
ITT, intention-to-treat population.
Biomarker evaluable population.
For the 23 low-MRD+ patients at EOT, 11 (47.8%) of 23 exhibited rising MRD before EOT, defined as at least a half log increase from the lowest MRD level for 2 consecutive visits during single-agent Ven treatment; the rest had stable MRD levels (supplemental Figure 7).
MRD clonal growth dynamic analysis
A total of 284 patients completed treatment without prior PD: 130 in the VenR arm and 154 in the BR arm. Patients with no MRD data (n = 23), with MRD data only on-treatment (n = 6), or only after PD/next line of treatment (n = 2), or with <2 measurable MRD values (n = 42), were excluded. This left a total of 211 patients (91 VenR treated and 120 BR treated) included in the population-based analysis. With a faster time to PD for the BR arm, median duration of MRD data collection post-EOT was 395 days for BR-treated patients and 735 days for VenR-treated patients.
The regrowth dynamics differed between treatment arms. Patients assigned to BR followed a typical logistic-type growth pattern overall (ie, growth curve approaches plateau); however, growth dynamics in the VenR arm largely followed an exponential growth pattern within the observation window.
Seventeen of 26 prognostic markers and patient demographics were tested as covariates (supplemental Table 2); treatment arm, IGHV status, TP53-mut status, risk of tumor lysis syndrome at study initiation (low/medium vs high), and age (<65 or ≥65 years) were identified as covariates on MRD growth rate (Table 2), whereas treatment arm, response status, and TP53-mut status were identified as covariates on MRD level at EOT. After considering other covariates, the effect of treatment arm on MRD growth rate and MRD level at EOT remained statistically significant: MRD growth rate for the VenR arm was 0.51-fold that of the BR arm (95% CI, 0.41, 0.64); MRD level at EOT for the VenR arm was 0.094-fold that for the BR arm (95% CI, 0.034, 0.266).
Median MRD level at EOT and median MRD doubling time post-EOT according to biological factors
| Parameter | VenR (n = 91) | BR (n = 120) | ||
|---|---|---|---|---|
| Median MRD at EOT | mut-IGHV (n = 22) | unmut-IGHV (n = 69) | mut-IGHV (n = 33) | unmut-IGHV (n = 87) |
| 3.40 × 10−5 | 1.88 × 10−5 | 1.11 × 10−3 | 4.46 × 10−4 | |
| P = .79 | P = .6 | |||
| TP53-WT* (n = 73) | TP53-mut* (n = 18) | TP53-WT* (n = 98) | TP53-mut* (n = 22) | |
| 1.87 × 10−5 | 3.56 × 10−5 | 1.94 × 10−2 | 3.07 × 10−2 | |
| P = .48 | P = .002 | |||
| Median MRD doubling time, d | mut-IGHV (n = 22) | unmut-IGHV (n = 69) | mut-IGHV (n = 33) | unmut-IGHV (n = 87) |
| 192 | 80 | 57 | 52 | |
| P = .0031 | P = .093 | |||
| TP53-WT* (n = 73) | TP53-mut* (n = 18) | TP53-WT* (n = 98) | TP53-mut* (n = 22) | |
| 101 | 66 | 54 | 45 | |
| P = .0012 | P = .072 | |||
| Age ≥65 y (n = 44) | Age <65 y (n = 47) | Age ≥65 y (n = 75) | Age <65 y (n = 45) | |
| 109 | 80 | 57 | 43 | |
| P = .012 | P = .0036 | |||
| Low/medium TLS risk (n = 65) | High TLS risk (n = 26) | Low/medium TLS risk (n = 86) | High TLS risk (n = 34) | |
| 105 | 63 | 56 | 51 | |
| P = .0001 | P = .02 | |||
| Parameter | VenR (n = 91) | BR (n = 120) | ||
|---|---|---|---|---|
| Median MRD at EOT | mut-IGHV (n = 22) | unmut-IGHV (n = 69) | mut-IGHV (n = 33) | unmut-IGHV (n = 87) |
| 3.40 × 10−5 | 1.88 × 10−5 | 1.11 × 10−3 | 4.46 × 10−4 | |
| P = .79 | P = .6 | |||
| TP53-WT* (n = 73) | TP53-mut* (n = 18) | TP53-WT* (n = 98) | TP53-mut* (n = 22) | |
| 1.87 × 10−5 | 3.56 × 10−5 | 1.94 × 10−2 | 3.07 × 10−2 | |
| P = .48 | P = .002 | |||
| Median MRD doubling time, d | mut-IGHV (n = 22) | unmut-IGHV (n = 69) | mut-IGHV (n = 33) | unmut-IGHV (n = 87) |
| 192 | 80 | 57 | 52 | |
| P = .0031 | P = .093 | |||
| TP53-WT* (n = 73) | TP53-mut* (n = 18) | TP53-WT* (n = 98) | TP53-mut* (n = 22) | |
| 101 | 66 | 54 | 45 | |
| P = .0012 | P = .072 | |||
| Age ≥65 y (n = 44) | Age <65 y (n = 47) | Age ≥65 y (n = 75) | Age <65 y (n = 45) | |
| 109 | 80 | 57 | 43 | |
| P = .012 | P = .0036 | |||
| Low/medium TLS risk (n = 65) | High TLS risk (n = 26) | Low/medium TLS risk (n = 86) | High TLS risk (n = 34) | |
| 105 | 63 | 56 | 51 | |
| P = .0001 | P = .02 | |||
TLS, tumor lysis syndrome.
TP53 status only [not considering del(17p)].
The model predicted that median MRD level at EOT was significantly lower post-VenR (1.88 × 10−5) vs post-BR (7.06 × 10−4; P = 5.1 × 10−8) (Figure 5A); median MRD doubling time post-EOT was significantly longer for patients treated with VenR (93 days; n = 91) vs BR (53 days; n = 120; P = 1.2 × 10−7) (Figure 5B). In the VenR arm, no significant difference in median MRD level at EOT was seen regardless of mut-IGHV or TP53-mut status (Table 2). However, in the BR arm, patients with TP53-mut had significantly higher median MRD levels at EOT than those with TP53-WT. For the combined treatment arms, median MRD doubling time was 53 days in patients aged <65 years (n = 92) and 66 days in patients aged ≥65 years (n = 119; P = .013); similar differences were seen in both treatment arms.
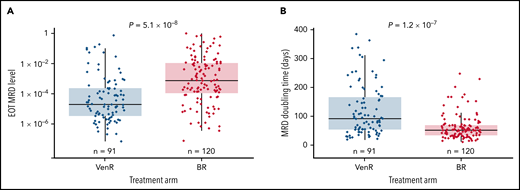
MRD level at EOT according to treatment arm (A) and MRD doubling time according to treatment arm (B). Analysis was unadjusted for covariates.
MRD level at EOT according to treatment arm (A) and MRD doubling time according to treatment arm (B). Analysis was unadjusted for covariates.
Multivariate Cox analysis showed that achieving uMRD at EOT and having mut-IGHV disease were independently associated with reduced risk of relapse (supplemental Table 3).
Response to next-line therapy
After a median treatment-free interval of 23.7 months (range, 3.3 to 43.8 months), 67 (77.0%) of 87 patients in the VenR arm and 123 (83.1%) of 148 patients in the BR arm who experienced PD received a subsequent therapy. Of the patients in the VenR arm who received subsequent therapy, 32 (47.8%) received a Ven-based therapy (21 within the MURANO substudy and 11 outside of the substudy), 18 (26.9%) received a Bruton’s tyrosine kinase inhibitor (BTKi), 15 (22.4%) received chemoimmunotherapy, and 2 (3.0%) received another novel agent. Of the patients in the BR arm who received a subsequent therapy, 72 (58.5%) received a BTKi, 24 (19.5%) received chemoimmunotherapy, 15 (12.2%) received a Ven-based therapy (9 within the MURANO substudy and 6 outside of the substudy), and 12 (9.8%) received another novel agent.
Response rates to subsequent treatment with Ven-based or BTKi-based therapy are shown in supplemental Table 4. In patients from the VenR arm who received subsequent treatment, best overall response rate (ORR) to Ven-based therapy was 72.2% (CR/CR with incomplete bone marrow [BM] recovery, 5.6%; ORR in patients with unmut-IGHV, 70.6% [n = 12 of 17]), and best ORR to BTKi-based therapy was 100.0% (CR/CR with incomplete BM recovery, 7.1%; ORR in patients with unmut-IGHV, 100% [n = 8 of 8]).
Safety
No new safety signals were identified. Excluding non-melanoma skin cancers, 2 additional second primary malignancies in the VenR arm (acute myeloid leukemia and plasma cell myeloma) were reported since the previous update. Rates of Richter’s transformation remained balanced between treatment arms (7 cases in the VenR arm and 6 in the BR arm) (supplemental Table 5).
Discussion
This 5-year update of MURANO found that survival benefits of VenR treatment over BR are maintained 3 years’ posttreatment cessation in patients with R/R CLL.
Continued clinical benefit with the fixed-duration VenR treatment strategy was shown. Of note, in patients assigned to 2 years of Ven treatment, median PFS at 3 years’ posttreatment cessation was particularly promising at 53.6 months. This was markedly better than the 17.0-month median PFS achieved in the BR arm and, to our knowledge, is superior to any PFS previously reported for chemoimmunotherapy in an R/R population. In the patients who received VenR, a substantial proportion who were uMRD with no PD at EOT maintained uMRD status (38.6%), and they continue to display a durable ongoing response. In the current analysis, MRD level continued to be a robust predictor of PFS: almost all patients with high-MRD+ status at EOT had PD before 3 years’ post-EOT, and many patients with uMRD at EOT continued to be in clinical remission 3 years’ post-EOT. uMRD status at EOT was also predictive of improved OS. These data are congruent with previous analyses.5,29
The advent of novel targeted agents along with the use of combination therapy in CLL has led to improved outcomes, which in the short term can be difficult to quantify. Similar to our findings, uMRD in CLL has been shown to correlate with prolonged PFS, and in some cases OS,3,7,36,37 suggesting that uMRD may be a useful surrogate end point for response/survival in clinical trials. Although there are currently no data to fully support MRD-directed treatment in CLL, the ongoing phase 2 CAPTIVATE trial (Phase 2 Study of the Combination of Ibrutinib Plus Venetoclax in Subjects With Treatment-naïve Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma) has shown that MRD assessment can be used to guide treatment discontinuation in patients with treatment-naive CLL.38 Further studies investigating the role of uMRD achievement in obtaining durable disease control may lead to MRD-based treatment regimens aiming for long-lasting MRD remission.
VenR continued to show an advantage over BR in PFS in all patient subgroups, including those with historically high-risk features such as del(17p) and/or TP53-mut. A similar trend was observed for OS, although median OS was NE for almost all subsets in both treatment groups. In the overall population, the OS benefits with VenR over BR were sustained from the 4-year update (HR, 0.41).5 Interestingly, in the first-line setting, Ven plus obinutuzumab (G) has been shown to achieve consistently high ORRs and CR rates, and improved PFS vs G-chlorambucil in patients with CLL, regardless of presence of unmut-IGHV, del(17p), or TP53-mut.28
unmut-IGHV, del(17p), and TP53-mut are clinically relevant biomarkers in predicting poor response to chemoimmunotherapy.4,24 In the current analyses, we showed that although these poor prognostic markers of CLL remain so for Ven-based regimens, outcomes are better relative to those with chemoimmunotherapy. In earlier analyses of MURANO, del(17p) and/or TP53-mut were associated with an increased risk of PD after Ven treatment cessation (P = .01 vs no deletion/mutation).29 In this 5-year update, unmut-IGHV, del(17p) and/or TP53-mut, and GC were all associated with significantly inferior PFS, regardless of treatment (P < .05 for all vs no high-risk factor) but with a longer PFS in the VenR arm vs BR. Similarly, OS was poorer among VenR-treated patients with del(17p) and/or TP53-mut vs patients with TP53-WT (P = .0059). In patients who received BR, despite inferior PFS among those with del(17p) and/or TP53-mut vs those without, OS was similar between the 2 groups, indicating that effective salvage therapies are now available and consistently used post-PD. Although in the VenR arm the unmut-IGHV subgroup had reduced 5-year OS vs the group with mut-IGHV (80.7% vs 92.3%, respectively), the difference was not significant (P = .876). Another marker indicative of a poor prognosis is deletion of chromosome 11q [del(11q)].39 Interestingly, patients treated with VenR in the current analysis had similar median PFS (54 months), regardless of del(11q) status. In the BR arm however, patients harboring del(11q) had a shorter median PFS (16 months) than those with normal 11q (22 months).
Patients with mut-IGHV have consistently superior PFS to those with unmut-IGHV after chemoimmunotherapy.20 Five-year follow-up of R/R CLL treated with the BTKi ibrutinib revealed a shorter median PFS among patients with unmut-IGHV vs mut-IGHV (43 and 63 months, respectively).40 Ven monotherapy has previously resulted in equivalently high ORRs in patients with (94%) or without (76%) mut-IGHV; however, this was with continuous therapy and shorter follow-up time (17 to 21 months).13 The inferior PFS observed here with time-limited treatment in patients with unmut-IGHV vs mut-IGHV seems attributable to the more rapid disease regrowth after treatment cessation. In the current study, patients from the VenR arm who experienced PD responded well to Ven retreatment, with a best ORR of 72.2% overall and 70.6% in patients with unmut-IGHV status (although patient numbers were small). These and other early data suggest that Ven retreatment retains efficacy in unmut-IGHV disease, opening the opportunity for prolonged benefit beyond a first progression event.41,42 Prolonged single-agent therapy is associated with emergence of BCL-2 resistance mutations,43,44 which may be mitigated by intermittent therapy. Optimization of Ven therapy use in an intermittent fashion will require further clinical trials.
GC and del(17p) were previously linked to poor PFS in MURANO, with patients with GC also exhibiting an increased frequency of high-MRD positivity at EOT and EOCT.5,29 In the current follow-up, patients with GC or del(17p) continued to have poorer survival outcomes after treatment than those with no GC or del(17p). unmut-IGHV, GC, and del(17p) were predictive of less favorable outcomes and independently associated with higher conversion rates to detectable MRD and subsequent PD in patients who had uMRD at EOT. In contrast, patients without del(17p) or GC, or with mut-IGHV, who achieved uMRD status at EOT were more likely to maintain uMRD status or experience MRD conversion without subsequent PD. Numbers in the GC or del(17p) biomarker subsets were small and could not be included in the MRD growth model; however, it is proposed that MRD doubling time would be more rapid in patients with these risk factors.
Similar to the trends observed with a Ven-based regimen in treatment-naive patients,32 we show here for the first time that MRD growth after VenR treatment (vs BR) in patients with R/R CLL is significantly slower, with MRD doubling time post-VenR therapy longer than post-BR (93 days vs 53 days, respectively). The slower growth kinetics with VenR treatment are reflected in clinical outcomes (eg, the longer TTNT in patients who received VenR vs BR).
Within the VenR arm, patients with unmut-IGHV or TP53-mut were able to achieve high rates of uMRD at EOT; however, consistent with the inferior clinical outcomes observed, these patients exhibited accelerated MRD doubling time post-EOT. Taken together, and considering the similar findings of the CLL14 trial (A Prospective, Open-Label, Multicenter Randomized Phase III Trial to Compare The Efficacy and Safety of A Combined Regimen of Obinutuzumab and Venetoclax Versus Obinutuzumab and Chlorambucil in Previously Untreated Patients With CLL and Coexisting Medical Conditions),32 EOT uMRD status in isolation among high-risk patients may be limited in predicting long-term clinical outcomes.
Conceptually, the logistic growth model comprised an initial exponential growth phase, followed by a slower growth phase when approaching the plateau (carrying capacity), making it an ideal model to describe the diverse trajectories of MRD regrowth. Although there was no plateau in MRD level observed in the VenR arm (possibly due to the shorter follow-up duration relative to the slower growth rate), this was not expected to affect the derivation of MRD doubling time, as the MRD values had already doubled within the observation window. Also, patients who were included in the clonal growth model were restricted to those whose disease had not progressed before EOT and who had ≥2 MRD measurements post-EOT; this may have led to a selection bias in the doubling time analysis due to exclusion of patients with the most adverse disease biology. This finding is consistent with the phase 3 CLL14 trial, in which median MRD doubling time was significantly longer after VenG therapy than after G-chlorambucil therapy in treatment-naive patients with CLL (MRD doubling time, 84 days vs 67 days, respectively; P = 3.3 × 10−5).32 The biology behind the slower disease re-growth after Ven-based therapy (vs cytotoxic chemoimmunotherapy) is unknown but could reflect lower rates of clonal evolution, less therapy-induced genomic instability, or disease clearance in multiple compartments (PB and BM); these hypotheses need further investigation.
There were no next-generation sequencing MRD data available for deeper assessments to the <10−5 or <10−6 cutoff, and BM MRD was not assessed due to the impracticality of serial BM analyses in a large multicenter trial. MRD results from PB and BM samples, however, have been reported to correlate in 90% to 95% of cases in which the uMRD cutoff is <10−4.6,9 As such, PB assessments were considered appropriate for evaluation of MRD kinetics.
A favorable safety profile for Ven was previously reported in MURANO,5,29,30 and the absence of any new safety signals or additional cases of Richter’s transformation during this 5-year follow-up is encouraging. Although there were 2 new secondary primary malignancies since the last update (excluding non-melanoma skin cancers), these AEs remained balanced across treatment arms, which has also been confirmed in an updated follow-up of the frontline CLL14 trial.32
The depth of remission and durable response with VenR treatment observed here is a promising finding. It should be noted, however, that >90% of the study population received chemoimmunotherapy as first-line treatment, with only 8 patients having had prior therapy with BTKis.30 Novel agents targeting the B-cell receptor pathway are increasingly used in the first-line setting.45 Although 2 of the 3 patients in the VenR arm with prior BTKi exposure all responded to treatment (one CR and one partial response [both had uMRD status]; the third patient died before response assessment), further studies are required to determine the true effect in an R/R CLL study population previously treated with BTKis.
In this update of MURANO, sustained survival benefits are shown with VenR over BR, up to 3 years’ post-completion of fixed-duration treatment and all patients off-therapy, regardless of the presence of high-risk cytogenetic biomarkers. uMRD at EOT with VenR was associated with improved PFS and OS, and unmut-IGHV, del(17p), and GC were associated with inferior outcomes in terms of MRD conversion and subsequent PD. Durable PFS was evident, and a substantial proportion of patients who completed VenR treatment retained uMRD status 3 years post-EOT, suggesting there is a patient subset who will achieve long-term disease control. These data further support the use of fixed-duration VenR in patients with R/R CLL.
Acknowledgments
The authors thank the patients and their families and all MURANO study team members and investigators. Special thanks are offered to Rosemary Harrup, Naomi Chang, Othman Al-Sawaf, Kirsten Fischer, and Can Zhang for their contributions.
Ven is being developed in a collaboration between Genentech, Inc. and AbbVie. Genentech, Inc. and AbbVie provided financial support for the study and participated in the design, study conduct, analysis, and interpretation of data, as well as the writing, review, and approval of the manuscript. Third-party medical writing assistance under the direction of A.P.K. and J.F.S. was provided by Sinéad Holland and Simon Lancaster of Ashfield MedComms, an Ashfield Health company, and was funded by F. Hoffmann–La Roche Ltd.
Authorship
Contribution: J.F.S., T.J.K., J.Q.W., and A.P.K. designed the study; J.F.S., T.J.K., J.d.l.S., and A.P.K. conducted the study; J.F.S., T.J.K., B.F.E., J.D., C.J.O., S.A., N.L., T.R., J.d.l.S., U.J., G.C., M.M., and A.P.K. conducted the recruitment and follow-up of patients; J.F.S., T.J.K., B.F.E., J.D., S.A., T.R., J.d.l.S., U.J., M.M., C.M., A.P., T.L., J.Q.W., Y.J., M.L., M.B., and A.P.K. collected data; J.F.S., T.J.K., B.F.E., J.D., S.A., T.R., J.d.l.S., U.J., M.M., C.M., B.C., A.P., T.L., J.Q.W., Y.J., M.L., M.B., and A.P.K. carried out data analysis; and J.F.S., T.J.K., B.F.E., J.D., C.J.O., S.A., N.L., T.R., J.d.l.S., U.J., G.C., M.M., B.C., T.L., J.Q.W., Y.J., M.L., M.B., and A.P.K. carried out data analysis. All authors read and approved the submitted version of the manuscript.
Conflict-of-interest disclosure: J.F.S. reports research funding from F. Hoffmann–La Roche Ltd., AbbVie, AstraZeneca, Pharmacyclics, Janssen, Genentech, Inc.; and honoraria from AbbVie, AstraZeneca, Janssen, Roche, Merck, Servier, Incyte, and Gilead. T.J.K. reports stock in and patents/royalties from Oncternal Therapeutics Inc.; research funding from Pharmacyclics, AbbVie, Breast Cancer Research Foundation, MD Anderson Cancer Center, Oncternal Therapeutics Inc., Specialized Center of Research (SCOR)–The Leukemia and Lymphoma Society, California Institute for Regenerative Medicine, National Cancer Institute/National Institutes of Health; and honoraria from Pharmacyclics, AbbVie, Genentech, Inc., F. Hoffmann–La Roche Ltd., Janssen, Gilead, National Cancer Institute/National Institutes of Health, and Celgene. B.F.E. reports consultancy for the Consultant Department I for Internal Medicine; research funding from Janssen, Gilead, F. Hoffmann–La Roche Ltd., AbbVie, BeiGene, and AstraZeneca; speakers bureau from Janssen, Gilead, F. Hoffmann–La Roche Ltd., AbbVie, Novartis, Celgene, Adaptive Biotechnologies, BeiGene, AstraZeneca, and Hexal; membership on an entity’s Board of Directors or advisory committees for Janssen, F. Hoffmann–La Roche Ltd., Novartis, AbbVie, Gilead, Celgene, ArQule, AstraZeneca, Oxford Biomedica (UK), BeiGene, and MSD; and travel, accommodations, and expenses from Janssen, F. Hoffmann–La Roche Ltd., Novartis, AbbVie, Gilead, and Celgene. J.D. reported honoraria from F. Hoffmann–La Roche Ltd.; and membership on an entity's Board of Directors or advisory committees for F. Hoffmann–La Roche Ltd. and AbbVie. C.J.O. reports research funding from F. Hoffmann–La Roche Ltd., AbbVie, AstraZeneca, Pharmacyclics, Janssen, and Genentech, Inc.; and honoraria from AbbVie, AstraZeneca, Janssen, F. Hoffmann–La Roche Ltd., Merck, Servier, Incyte, and Gilead. S.A. reports consultancy and honoraria from AbbVie and Roche; speaker engagements for AbbVie; and research support from Roche. N.L. reports SAB, consultancy, honoraria from AbbVie, Adaptive Biosciences, AstraZeneca, BeiGene, Celgene, Genentech Inc., Janssen, LOXO/Eli Lilly, and Pharmacyclics; and institutional research funding from AbbVie, AstraZeneca, BeiGene, Genentech Inc., LOXO/Eli Lilly, MingSight, Octapharma, Oncternal, TG Therapeutics, and Verastem. T.R. reports consultancy for Sandoz, Janssen, AbbVie, F. Hoffmann–La Roche Ltd., and Gilead; research funding from AbbVie, Pharmacyclics, Gilead, GlaxoSmithKline, Novartis, Janssen, F. Hoffmann–La Roche Ltd., Acerta, AstraZeneca, BioGene, and UCB; and honoraria from AbbVie, Pharmacyclics, Novartis, Janssen, Acerta, AstraZeneca, BioGene, UCB, and Sandoz. J.d.l.S. reports consultancy for AbbVie, AstraZeneca, Janssen, and F. Hoffmann–La Roche Ltd.; research funding from AbbVie, AstraZeneca, Pharmacyclics, F. Hoffmann–La Roche Ltd., and TG Therapeutics; and speakers bureau, membership on an entity's Board of Directors, or advisory committees for AbbVie, AstraZeneca, and Janssen. U.J. reports consultancy for Novartis, Gilead, BMS/Celgene, Janssen, and Annexon; and honoraria and advisory boards for AbbVie, Roche, Amgen, Gilead, BMS/Celgene, Janssen, Novartis, Miltenyi, and Incyte. G.C. reports consultancy for Roche and Celgene; advisory board for MedXcell, MabQi, and Ownards Therapeutics; and honoraria from AbbVie, Incyte, Gilead, Novartis, Jansen, Roche, and Celgene. M.M. reports advisory board and honoraria from AbbVie, Acerta/AstraZeneca, and F. Hoffmann–La Roche Ltd.; honoraria from Janssen; and advisory board for Verastem. C.M. reports employment with Genome Diagnostic Laboratory, Amsterdam University Medical Centre; consultancy with cytogenetic field; and research funding in financial support related to microarray analysis of MURANO samples. B.C. reports employment and shares in AbbVie. A.P., M.L., and M.B. report employment and shares in F. Hoffmann–La Roche Ltd. T.L. and Y.J. report employment and shares in Genentech, Inc. J.Q.W. reports former employment with Genentech, Inc. A.P.K. reports employment with Amsterdam University Medical Centers, University of Amsterdam; research funding from AbbVie, Genentech, Inc./F. Hoffmann–La Roche Ltd., BMS, AstraZeneca, and Janssen; membership on an entity’s Board of Directors or advisory committee with AbbVie, Genentech, Inc./F. Hoffmann–La Roche Ltd., BMS, AstraZeneca, Janssen, and LAVA; and honoraria from AbbVie.
The current affiliation for A.P. is Janssen (Johnson & Johnson), High Wycombe, United Kingdom.
The current affiliation for J.Q.W. is Bristol Myers Squibb, South San Francisco, CA.
James D’Rozario died on 27 January 2022.
Correspondence: Arnon P. Kater, Department of Hematology, Cancer Center Amsterdam, Lymphoma and Myeloma Center Amsterdam, Amsterdam University Medical Centers, Meibergdreef 9, 1105 AZ Amsterdam, The Netherlands; e-mail: a.p.kater@amsterdamumc.nl.
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are also available (https://vivli.org/members/ourmembers/). Additional details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents can be found on the following: https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal