

Key Points
We use base editing to create 7CAR8, a quadruple-edited CART targeting CD7, designed for allogeneic use.
We demonstrate the efficacy of 7CAR8 for potential clinical translation for relapsed or refractory T-ALL and other CD7+ malignancies.

Abstract
Allogeneic chimeric antigen receptor T-cell (CART) therapies require multiple gene edits to be clinically tractable. Most allogeneic CARTs have been created using gene editing techniques that induce DNA double-stranded breaks (DSBs), resulting in unintended on-target editing outcomes with potentially unforeseen consequences. Cytosine base editors (CBEs) install C•G to T•A point mutations in T cells, with between 90% and 99% efficiency to silence gene expression without creating DSBs, greatly reducing or eliminating undesired editing outcomes following multiplexed editing as compared with clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9). Using CBE, we developed 7CAR8, a CD7-directed allogeneic CART created using 4 simultaneous base edits. We show that CBE, unlike CRISPR-Cas9, does not impact T-cell proliferation, lead to aberrant DNA damage response pathway activation, or result in karyotypic abnormalities following multiplexed editing. We demonstrate 7CAR8 to be highly efficacious against T-cell acute lymphoblastic leukemia (T-ALL) using multiple in vitro and in vivo models. Thus, CBE is a promising technology for applications requiring multiplexed gene editing and can be used to manufacture quadruple-edited 7CAR8 cells, with high potential for clinical translation for relapsed and refractory T-ALL.
Introduction
Personalized immunotherapies such as autologous chimeric antigen receptor T (CART) cells have revolutionized the treatment of relapsed or refractory (r/r) B-cell malignancies, including B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma, and have led to the regulatory approvals of several products.1,2 However, the use of patient-derived autologous products is fraught with several issues, including challenges with harvesting sufficient healthy T cells from ill patients, extended vein-to-vein time resulting from drug product manufacturing and release, and the risk of contaminating T cells with cancer cells in patients with active malignancies.1,3
Universally compatible, allogeneic CART derived from healthy donors may have the potential to deliver an on-demand, standardized infusion product that overcomes the manufacturing limitations of autologous CART and mitigates product heterogeneity that contributes to variability in treatment efficacy. Recent progress has been made in developing clinical allogeneic CART to obviate the need for autologous products.4,5 However, in addition to genetic modifications that may be necessary to overcome technical limitations such as fratricide in instances where the target antigen is expressed on both malignant cells and healthy T cells, allogeneic CART require further modifications to prevent graft-versus-host disease (GVHD) and CART rejection by recipient immune cells. To overcome these barriers, many allogeneic CART genome-editing approaches use DNA double-strand break (DSB)-inducing nucleases such as zinc finger nucleases,6,7 transcription activator-like effector nucleases,5,8,9 megaTALs,10 homing endonucleases,11 or clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (CRISPR-Cas) nucleases such as Streptococcus pyogenes CRISPR-associated protein 9 (spCas9)12 to modify multiple genomic loci. However, DSBs result in unpredictable, undesirable outcomes including complex genomic rearrangements, megabase-scale deletions, and chromothripsis.13-15 Simultaneous induction of multiple DSBs can further result in high frequency translocations between the on-target sites, capable of persisting in patients for at least several months.5,16 On-target genomic rearrangements resulting from DSBs have potential implications for the clinical use of nuclease-edited CART, and the potential for unintended on-target editing outcomes have been investigated in the pausing of a clinical trial (#NCT04416984; clinicaltrials.gov).
Base editing is an emerging genome editing technology capable of creating programmable single base pair changes at defined genomic loci with high precision and efficiency.17-19 Adenine base editors (ABEs) and cytosine base editors (CBEs) combine a single-stranded DNA deaminase enzyme with a nickase, spCas9, to install A•T to G•C or C•G to T•A point mutations at genomic target sites, respectively. CBE further incorporates 2 uracil glycosylase inhibitor domains to enhance the efficiency of C•G to T•A editing by transiently inhibiting base excision repair.20 Because both ABEs and CBEs operate without creating DSBs, multiplexed editing with either technology may result in efficient on-target editing and minimal undesired editing outcomes compared with nuclease editing. Both ABEs and CBEs are capable of disrupting gene expression by introducing point mutations in canonical splicing motifs or start codons; however, CBEs can also install premature termination codons in genes and thus may have broader targeting range for gene silencing applications.
To determine if CBE is clinically tractable, we developed an allogeneic CART for r/r T-cell acute lymphoblastic leukemia (T-ALL). Autologous CART have encountered limitations in treating T-ALL due to fratricide and the potential for disease contamination in patient-derived T cells.21 Moreover, patients with r/r disease tend to be chemorefractory, thus making it challenging to obtain remission for collection of autologous T-cell products.22 The use of allogeneic CART is further complicated by the associated risks of graft rejection and GVHD.23 The development of allogeneic CART for T-ALL therefore requires multiple genetic modifications, making T-ALL–directed CART an optimal candidate for CBE.
A key immunotherapy target previously identified for T-ALL is the surface receptor CD7, which is highly expressed on the vast majority of T-ALL blasts.24-28 However, CD7 is also present on most healthy T cells, and attempts by other groups to manufacture anti-CD7 CART without silencing expression of CD7 resulted in significant fratricide.14 Although CD7 is a costimulatory molecule, loss of CD7 expression is well tolerated by T cells, and anti-CD7 CART using nuclease-mediated disruption of CD7 expression have been shown to be effective against T-ALL in vitro and in vivo.25,26 Recently, HLA-matched donor-derived CART targeting CD7 in adult and pediatric T-ALL achieved an impressive complete remission rate in 18 of 20 treated patients.4 However, the use of HLA-matched donor-derived cells, although compelling, has significant practical challenges as it requires the rapid collection of T cells from healthy HLA-matched persons for individualized product manufacture. Due to the nature of T-ALL and the technical limitations associated with producing individualized autologous products for these patients, allogeneic anti-CD7 CART are a desirable potential treatment option.
We report herein on the use of CBE to develop a clinically compatible quadruple-base-edited allogeneic CART targeting CD7 (7CAR8) and preclinical testing in childhood T-ALL models.17 To our knowledge, 7CAR8 is the first CART with 4 simultaneous genetic edits progressing toward clinical development. In contrast to multiplexed editing of T cells with spCas9 messenger RNA (mRNA), we demonstrate that multiplexed editing of T cells with CBE mRNA does not impact T-cell proliferation, result in detectable translocations or karyotypic abnormalities, or lead to increased expression of genes involved in DNA damage or proapoptotic pathways. We provide robust preclinical data in patient-derived xenograft (PDX) models, establishing that 7CAR8 may be a highly effective therapeutic option for treating T-ALL and that CBE can be used to produce clinically relevant, good manufacturing practice-compliant 7CAR8 products with substantially lowered risk of genomic rearrangements or translocations.
Methods
Evaluation of cytokine-independent growth
Cells were plated at 6.67 × 105 cells per mL on day 0 in complete media with or without 300 IU/mL interleukin-2 (IL-2). Viability was assayed by flow cytometry using 7-actinomycin D staining. Half of the culture medium was removed on days 4, 7, 11, and 14 and the volume replaced by fresh media of the corresponding type. On day 18, cell viability was assayed as on day 0. Final cell counts were adjusted to compensate for the interim dilutions. The expansion factor of 7CART cells with and without exogenous IL-2 in culture was calculated by cell count and viable cell density at day 18 relative to day 0 of the assay. An expansion factor >1 represents cell proliferation during the course of the assay.
Next-generation sequencing (NGS) of genomic DNA samples Genomic DNA samples were prepared using QuickExtract DNA Extraction Solution. Genomic DNA was extracted from 5 × 105 T cells in 100 µL of QuickExtract according to the manufacturer’s protocol. Two microliters of extracted genomic DNA solution was added to a 25 µL polymerase chain reaction (PCR) mixture containing Q5 High-Fidelity DNA Polymerase and a 0.5 µM concentration of each forward and reverse primer. Following PCR amplification, PCR products were amplified using Illumina barcoding primers. Barcoding PCRs were performed using Q5 High-Fidelity DNA Polymerase and contained a 0.5 µM concentration of each forward and reverse barcoding primer and 2 µL of PCR mixture containing the amplified genomic site of interest in a total volume of 25 µL. Primers used for genomic DNA amplification are listed in supplemental Table 9. Following barcoding, PCR samples were purified using Solid Phase Reversible Immobilization beads and quantified using a NanoDrop 1000 Spectrophotometer, and DNA was sequenced on an Illumina MiSeq instrument.
RNA purification and sequencing
To investigate potential guide RNA-independent off-target RNA editing, total RNA was isolated from untreated and edited cell samples using the Quick-RNA Miniprep Plus Kit (Zymo Research). After RNA concentration quantification and the RNA quality evaluation, 400 ng of total RNA from each sample was subjected to mRNA isolation and strand-specific RNA sequencing library preparation using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England BioLabs). The RNA sequencing libraries were sequenced on an Illumina HiSeq X instrument.
Analysis of whole-transcriptome and next-generation sequencing
All targeted NGS data were analyzed as previously described.29 Briefly, NGS data were analyzed by performing 4 general steps: (1) Illumina demultiplexing, (2) read trimming and filtering, (3) alignment of all reads to the expected amplicon sequence, and (4) generation of alignment statistics and quantification of editing rates.
To analyze differential gene expression, paired-end FASTQ files were aligned to the human genome (Gencode GRCh38v31) using STAR (v2.7.2a) with parameters set to specify the ReadGroup and output both a genome-aligned BAM file and a transcriptome-aligned BAM file. Transcript abundance was quantified using RSEM (v1.3.1). Differential gene expression analysis was done with the Bioconductor package edgeR using a false discovery rate cutoff of 5% for determining differentially expressed genes.
Cell lines
CCRF-CEM cells stably expressing the green fluorescent protein (GFP) firefly luciferase transgene were obtained from the laboratory of Maksim Mamonkin at Baylor College of Medicine.30
Cytotoxicity assay
Antitumor cytotoxicity of 7CAR8 cells was assessed using an image-based coculture method with the T-ALL cell line CCRF-CEM cells modified to stably express GFP. Tumor cells were plated in a tissue culture–treated 96-well plate and allowed to adhere for 24 hours. 7CAR8 cells or untransduced T cells (UTD) were then introduced to the tumor cells at 4:1, 2:1, and 1:1 effector-to-tumor ratios. Coculture plates were incubated in the Incucyte Live-Cell Analysis System (Sartorius, Göttingen, Germany) for up to 120 hours to measure the killing of GFP expressing CCRF-CEM tumor cells.
Multicytokine-release assays
Interferon γ (IFN-γ), IL-2, and tumor necrosis factor α were measured by enzyme-linked immunosorbent assay using the Ella platform (ProteinSimple, San Jose, CA). 7CAR8 cells (0.25 × 106) were cocultured in a 1:1, 1:2, and 1:4 ratio with Dynabeads (Invitrogen, Waltham, MA) coupled to recombinant human CD7 (ACRO Biosystems, Newark, DE) for 18 to 24 hours. Supernatants were collected and analyzed for production of each cytokine.
7CAR8 IFN-γ assay
CAR-mediated IFN-γ production is measured by enzyme-linked immunosorbent assay using the Ella platform (ProteinSimple). 7CAR8 drug product cells (0.5 × 106) were cultured with 100 µg Dynabeads (Invitrogen) coupled to recombinant human CD7 (ACROBiosystems) for 18 to 24 hours. Supernatants were collected and analyzed for levels of IFN-γ.
IsoLight polyfunctionality assay
Polyfunctionality was assessed using the single-cell secretome platform on the IsoLight system (IsoPlexis, Branford, CT). 7CAR8 cells (1 × 106) were cultured in 24-well plates coated with recombinant human CD7 (ACROBiosystems) for 18 to 24 hours. After stimulation, cells were collected, stained with a cell-membrane antibody cocktail, loaded onto the IsoCode single-cell chip, and then analyzed on the IsoLight system.
Patient samples
Patient samples were collected from children with newly diagnosed T-ALL after informed consent was obtained. Patient samples were viably cryopreserved. Informed consent for use of specimens for further research was obtained in accordance with the Declaration of Helsinki. A subset of patients had early T-cell precursor (ETP) ALL, a biologically distinct subtype of T-ALL associated with a poor initial response to chemotherapy.
Flow cytometry
All PDX were assessed for surface expression of CD7, CD45, PD1, and its ligand PD-L1. Antibodies were obtained from Miltenyi, and data were acquired using a FACS Verse flow cytometer. CAR expression and base-editing efficiencies were assessed using flow cytometry as described in supplemental Table 10. Data were analyzed using FlowJo version 10.8 (BD Biosciences, Franklin Lakes, NJ). A cutoff of 20% positivity was used as a binary criterion for positivity of CD7 expression on PDX screening.
Statistical analyses
General statistical analyses were performed in R (version 4.0.4) using RStudio (RStudio, PBC, Boston, MA).31 Survival analyses were performed in Prism (version 9). Survival curves were compared using the log-rank test. Bonferroni test was applied for multiple comparisons where appropriate. To test for overrepresentation of gene ontology (GO) terms in up or down differentially expressed genes, a GO enrichment analysis using a linear model fit was done with the Bioconductor edgeR function goann. Results for gene ontologies were limited to those belonging to the biological process ontology. Annotation maps describing the human gene ontology were obtained using the Bioconductor package GO.db.
See supplemental Methods for more details.
Results
Base editing is a highly efficient strategy for T-cell genomic editing
CBEs can be used to convert C•G base pairs to T•A base pairs in the DNA encoding a protein of interest to install either a premature termination codon or mutate conserved dinucleotide motifs present at exon-intron boundaries to disrupt mRNA splicing (Figure 1A). Both of these base editing strategies efficiently alter protein expression. To determine whether CBE can introduce simultaneous multiplexed edits, we edited primary human T cells at 1, 2, or 3 target sites simultaneously using a single electroporation (EP) of mRNA encoding CBE and synthetic gRNAs targeting β-2-microglobulin (B2M), T-cell receptor α chain (TRAC), and programmed cell death protein 1 (PDCD1), the gene encoding PD1. The addition of gRNAs targeting up to 3 sites resulted in efficient on-target editing (72.0% to 96.2%) without impairing the efficiency of any one edit (Figure 1B).
![Base editing is a highly efficient alternative to nuclease editing. (A) The efficiency of base editing measured by NGS amplicon sequencing in percentage of C-to-T edits at the targeted site is shown on the y-axis. The efficiency at each of 3 sites is demonstrated: β-2 microglobulin (B2M), T-cell receptor α chain (TRAC), and programmed cell death protein 1 (PDCD1). High efficiency is maintained with multiple edits. (*P < .05, all other comparisons not significant). (B) A comparison of CBEs with spCas9. We demonstrate that when 2 or 3 edits are made with CBE, T-cell yield is not impacted (teal bars). In contrast, when spCas9 is used, T-cell yield decreases in a manner proportional to the number of edits made (gold bars). X-axis depicts number of targets edited, and y-axis depicts percentage cell yield as compared with the EP-only condition (*P < .05, **P < .005, all other comparisons not significant). Statistical testing in (A) and (B) was performed using 2-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples. (C) UDiTaS was used to measure translocation frequencies between on-target editing sites in T cells from 1 donor edited at 3 target sites simultaneously. spCas9 induced translocations at on-target sites with frequencies between 0.5% and 1.6%, whereas CBE did not induce detectable translocations. (D) Volcano plots of differentially expressed genes between T cells edited with CBE and Cas9 identified through whole-transcriptome RNA sequencing. Orange circles represent genes that are upregulated and green circles represent genes that are downregulated following editing. Red circles represent genes that are differentially expressed and part of the tumor suppressor TP53 pathway. SpCas9-edited T cells significantly upregulated the TP53 pathway, and CBE does not. The x-axis represents log2 (fold change [FC]), and the y-axis represents P adjusted with the Benjamini-Hochberg method to control for false discovery rate. Significance was determined by an adjusted P value ≤0.05.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/6/10.1182_blood.2022015825/3/m_bloodbld2022015825f1.png?Expires=1767736286&Signature=zBMQHvVe5SMAUWgNH5sHeKnt53BojR4UA7VEIWE18BIC1RczRMtz3wlFgz47-yLmv2aeQc-ztCcR3YfIgABRU~vRRRiPyIPpILoPHXiDLhgfzsANi-Mai6PmHbKxqA2UB0~FLZm5G8pc1t3DftJQKghpNTr9CCQK9YwU7ld13QfFx5OE46CpHQaU37cXiCL9sBh4YqWzlk2wU9sI4t~0SyuimpKoLks~XJKmbbnEf~Z8ExFzS2RqR56E8s3TZ1LeXTrhatE8Nv8Uz3L0axF9g6uKIe8kclP9gqq2eesAdK0nTdzsNRqhYz94xgxv4128YpZkBfdoY2NNNN5FwV1DCg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Base editing is a highly efficient alternative to nuclease editing. (A) The efficiency of base editing measured by NGS amplicon sequencing in percentage of C-to-T edits at the targeted site is shown on the y-axis. The efficiency at each of 3 sites is demonstrated: β-2 microglobulin (B2M), T-cell receptor α chain (TRAC), and programmed cell death protein 1 (PDCD1). High efficiency is maintained with multiple edits. (*P < .05, all other comparisons not significant). (B) A comparison of CBEs with spCas9. We demonstrate that when 2 or 3 edits are made with CBE, T-cell yield is not impacted (teal bars). In contrast, when spCas9 is used, T-cell yield decreases in a manner proportional to the number of edits made (gold bars). X-axis depicts number of targets edited, and y-axis depicts percentage cell yield as compared with the EP-only condition (*P < .05, **P < .005, all other comparisons not significant). Statistical testing in (A) and (B) was performed using 2-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples. (C) UDiTaS was used to measure translocation frequencies between on-target editing sites in T cells from 1 donor edited at 3 target sites simultaneously. spCas9 induced translocations at on-target sites with frequencies between 0.5% and 1.6%, whereas CBE did not induce detectable translocations. (D) Volcano plots of differentially expressed genes between T cells edited with CBE and Cas9 identified through whole-transcriptome RNA sequencing. Orange circles represent genes that are upregulated and green circles represent genes that are downregulated following editing. Red circles represent genes that are differentially expressed and part of the tumor suppressor TP53 pathway. SpCas9-edited T cells significantly upregulated the TP53 pathway, and CBE does not. The x-axis represents log2 (fold change [FC]), and the y-axis represents P adjusted with the Benjamini-Hochberg method to control for false discovery rate. Significance was determined by an adjusted P value ≤0.05.
Base editing is a highly efficient alternative to nuclease editing. (A) The efficiency of base editing measured by NGS amplicon sequencing in percentage of C-to-T edits at the targeted site is shown on the y-axis. The efficiency at each of 3 sites is demonstrated: β-2 microglobulin (B2M), T-cell receptor α chain (TRAC), and programmed cell death protein 1 (PDCD1). High efficiency is maintained with multiple edits. (*P < .05, all other comparisons not significant). (B) A comparison of CBEs with spCas9. We demonstrate that when 2 or 3 edits are made with CBE, T-cell yield is not impacted (teal bars). In contrast, when spCas9 is used, T-cell yield decreases in a manner proportional to the number of edits made (gold bars). X-axis depicts number of targets edited, and y-axis depicts percentage cell yield as compared with the EP-only condition (*P < .05, **P < .005, all other comparisons not significant). Statistical testing in (A) and (B) was performed using 2-tailed Student’s t-test according to the method of Benjamini, Krieger, and Yekutieli without assuming equal variances between samples. (C) UDiTaS was used to measure translocation frequencies between on-target editing sites in T cells from 1 donor edited at 3 target sites simultaneously. spCas9 induced translocations at on-target sites with frequencies between 0.5% and 1.6%, whereas CBE did not induce detectable translocations. (D) Volcano plots of differentially expressed genes between T cells edited with CBE and Cas9 identified through whole-transcriptome RNA sequencing. Orange circles represent genes that are upregulated and green circles represent genes that are downregulated following editing. Red circles represent genes that are differentially expressed and part of the tumor suppressor TP53 pathway. SpCas9-edited T cells significantly upregulated the TP53 pathway, and CBE does not. The x-axis represents log2 (fold change [FC]), and the y-axis represents P adjusted with the Benjamini-Hochberg method to control for false discovery rate. Significance was determined by an adjusted P value ≤0.05.
We next evaluated the impact of editing using spCas9 or CBE on T-cell yield using the same gRNAs for spCas9 or CBE editing (Figure 1C). Edited, untransduced cells were used in this comparison to avoid the introduction of confounding variables that may result from CAR signaling. Simultaneous editing at 2 or 3 sites using spCas9 resulted in 24.4% and 41.4% reduced T-cell yield, respectively, whereas a single site edit did not impact cell yield. In contrast, T-cell yield was unaffected by simultaneous CBE editing at up to 3 sites as compared with an EP-only control sample. Because targeted insertion of a CAR transgene can result in a more homogeneous cell therapy with potentially improved functionality,12 we further demonstrated that base editing can be multiplexed with targeted gene insertion at the TRAC locus using the Cas12b nuclease to enable complex genetic modifications of T cells while limiting the deleterious on-target editing effects of nuclease-mediated gene editing (supplemental Figure 1).32
We hypothesized that impaired cell yield in spCas9-treated T cells may result from the development of genomic rearrangement products and the activation of DNA damage response pathways in response to simultaneous induction of multiple DSBs. To test this, we determined the frequency of translocations and karyotypic abnormalities resulting from multiplexed spCas9 or CBE editing. T cells from one donor that were edited at 3 target sites simultaneously were evaluated using UDiTaS, and translocations between all on-target sites were identified with frequencies ranging between 0.4% to 1.6% in spCas9-treated cells but were not found in CBE-treated cells (Figure 1D).33 To select the optimal gRNA to measure karyotypic abnormalities in our quadruple-edited CART, gRNAs targeting each of the 4 target genes were screened for on-target CBE genomic editing activity and ability to silence protein expression (supplemental Table 5). We next assessed the karyotypes of cells edited simultaneously with each of these 4 gRNAs and either spCas9 or CBE using G-banded karyotyping (supplemental Table 1). In this assay, 22 of 100 spCas9-treated cells exhibited aberrant karyotypes associated with the 4 on-target editing sites, whereas 0 of 100 cells treated with CBE had karyotypic abnormalities detectable at the on-target editing sites.
To determine the impact of multiplexed gene editing on global gene expression, we performed whole-transcriptome sequencing of T cells edited simultaneously at 4 loci with spCas9 or CBE 24 or 48 hours following EP compared with an EP-only control. SpCas9 editing resulted in statistically significant upregulation of 66 genes, including 23 genes involved in the TP53 pathway, and significant downregulation of 12 genes. Strikingly, editing with CBE resulted in decreased expression of CD7 and CD52, 2 of the 4 targeted genes, and no other statistically significant changes in gene expression (Figure 1D; supplemental Table 2). Although transcripts from CD52 and PDCD1 most likely were degraded through nonsense-mediated decay, the mRNA transcripts for CD7 and TRAC were stably expressed but with retained intronic sequences that result in abrogated protein expression. Gene ontology analysis of the transcriptome sequencing data demonstrated that many of the upregulated genes resulting from spCas9 editing were associated with pathways involved in cellular apoptosis, programmed cell death, and intrinsic apoptotic signaling through the TP53 gene pathway, similar to what has previously been reported in other contexts (supplemental Table 3).34,35 Taken together, the high rate of genomic rearrangements and activation of cellular pathways involved in apoptosis provide potential mechanisms for the observed impaired cell yield in multiplex spCas9-edited cells.
CBE can be used to manufacture CARTs in a GMP-compliant process
We next used CBE to create 7CAR8, a CART targeting CD7. 7CAR8 is engineered to contain 4 simultaneous base edits to prevent expression of the proteins CD52, CD7, PD1, and TCRα and concurrent lentiviral transduction to introduce the anti-CD7 CAR transgene. CD7 expression is silenced to avoid fratricide that would otherwise prevent CART manufacturing; T cell receptor α chain is silenced to greatly reduce the potential for 7CAR8 to cause GVHD; PD1 is silenced to reduce immune-mediated CART inhibition. Finally, because the recipient immune system is capable of robust elimination of allogeneic cells, CD52 is edited to enable the use of the anti-CD52 antibody alemtuzumab to be employed as part of the lymphodepletion regimen prior to 7CAR8 infusion. This allows for the eradication of competing normal host T and natural killer cells while sparing 7CAR8 cells and facilitating their activation and expansion in vivo. However, because most mature T cells are CD7+, it is possible that 7CAR8 may eliminate the recipient T-cell compartment and thus may not require alemtuzumab-mediated lymphodepletion to escape allorejection by recipient T cells, which could allow for successful clinical translation with reduced-intensity lymphodepletion regimens.36
Electroporation of mRNA-encoding CBE and 4 synthetic gRNAs resulted in highly efficient editing (96.0% to 99.5%) at all 4 target sites, as measured by NGS and flow cytometry, using cells isolated from 3 healthy donors in a clinical-scale process (Figure 2A). On average, 92.9% of all T cells had been base edited at the 4 intended target sites, and 81.4% of all T cells had all 4 base edits and expressed the CAR construct (Figure 2B,D; supplemental Table 4). Because the intended base edits disrupt canonical splice donor sequences, we employed whole-transcriptome RNA sequencing to determine the resulting mature mRNA outcomes resulting from expression of edited alleles. We found that splice donor sequence disruption led to variable outcomes, including non–sense-mediated decay, intron retention, or activation of cryptic splice donor sites (supplemental Table 5). Regardless of the outcome of the mature mRNA sequence, all on-target base edits resulted in complete abrogation of detectable surface protein expression. We further evaluated translocations between on-target editing sites in 7CAR8 cells compared with control EP cells and spCas9-edited cells using UDiTaS and found no enrichment of translocations above background in the CBE-treated cells. However, analysis of spCas9-treated cells resulted in detection of on-target translocations at efficiencies between 0.25% and 4.01% (Figure 2C). We employed a cytokine-independent growth assay to assess the potential for transformation of 7CAR8 cells resulting from the lentiviral transduction and multiplexed base editing and determined that 7CAR8 cells manufactured in 3 separate lots were unable to grow in the absence of exogenous IL-2 (supplemental Table 4).
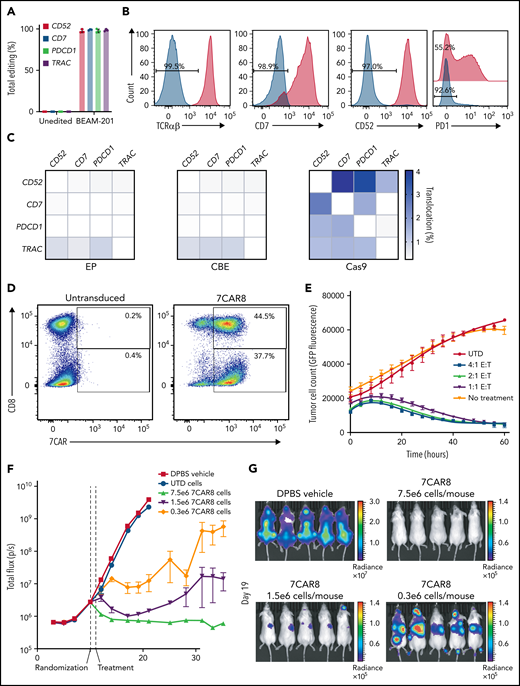
Base editing can be used to create a multiplexed edited CART modified at multiple loci. (A) NGS (left) and flow cytometry expression (right) of 4 proteins knocked out from the CART 7CAR8 using CBE. CD52, CD7, PDCD1, and TRAC were all knocked out with high efficiency. (B) Protein reduction measured using flow cytometry for the 4 editing targets. (C) Translocation analysis using the UDiTaS assay between the 4 on-target editing sites for EP-only control cells, CBE-edited cells, and spCas9-edited cells produced from 3 healthy donors. (D) Expression of CAR measured in CD8− and CD8+ T cells using flow cytometry in 7CAR8 and untransduced T cells. X-axis depicts CAR+ expression measured by staining with fluorophore-conjugated recombinant human CD7 protein. Y-axis depicts CD8 expression. (E) 7CAR8 cells produced from 3 separate donors cocultured with GFP-luciferase expressing T-ALL cell line CCRF-CEM in a 4:1, 2:1, and 1:1 E:T ratio. GFP expression over time was measured using the IncuCyte system. (F) Mice injected with the cell line CCRF-CEM modified to expressed GFP-luciferase were randomized to be treated with vehicle, untransduced T cells, or increasing doses of 7CAR8 (n = 10 mice per arm). A dose-dependent decrease in the surrogate disease marker total flux over time was seen in mice treated with 7CAR8. (G) Representative images of mice treated with vehicle and different doses of 7CAR8, with mice receiving the highest dose of 7CAR8 having no disease present.
Base editing can be used to create a multiplexed edited CART modified at multiple loci. (A) NGS (left) and flow cytometry expression (right) of 4 proteins knocked out from the CART 7CAR8 using CBE. CD52, CD7, PDCD1, and TRAC were all knocked out with high efficiency. (B) Protein reduction measured using flow cytometry for the 4 editing targets. (C) Translocation analysis using the UDiTaS assay between the 4 on-target editing sites for EP-only control cells, CBE-edited cells, and spCas9-edited cells produced from 3 healthy donors. (D) Expression of CAR measured in CD8− and CD8+ T cells using flow cytometry in 7CAR8 and untransduced T cells. X-axis depicts CAR+ expression measured by staining with fluorophore-conjugated recombinant human CD7 protein. Y-axis depicts CD8 expression. (E) 7CAR8 cells produced from 3 separate donors cocultured with GFP-luciferase expressing T-ALL cell line CCRF-CEM in a 4:1, 2:1, and 1:1 E:T ratio. GFP expression over time was measured using the IncuCyte system. (F) Mice injected with the cell line CCRF-CEM modified to expressed GFP-luciferase were randomized to be treated with vehicle, untransduced T cells, or increasing doses of 7CAR8 (n = 10 mice per arm). A dose-dependent decrease in the surrogate disease marker total flux over time was seen in mice treated with 7CAR8. (G) Representative images of mice treated with vehicle and different doses of 7CAR8, with mice receiving the highest dose of 7CAR8 having no disease present.
To assess the antigen-dependent reactivity of 7CAR8 and to evaluate the impact of silencing PDCD1 expression, we compared the cytokine secretion profiles of 7CAR8 cells with wild-type PDCD1 and 7CAR8 cells bearing the PDCD1 base edit in cells produced from 2 independent donors using an in vitro restimulation assay. Silencing PDCD1 expression increased polyfunctionality of the cell product manufactured from one donor and had no effect on 7CAR8 cells manufactured from a second donor (supplemental Figure 2). Because increased polyfunctionality of preinfusion CARTs is associated with improved clinical outcomes in other diseases, the PDCD1 edit was included in the final 7CAR8 product.37
In vitro and in vivo activity of 7CAR8 in T-ALL cell line models
We next assessed the efficacy of 7CAR8 in in vitro and in vivo preclinical models of T-ALL. To assess in vitro 7CAR8 cytotoxicity, the CD7+ T-ALL cell line CCRF-CEM, genetically modified to stably express a GFP-luciferase transgene,25 was cocultured with 7CAR8 cells in a 4:1, 2:1, and 1:1 effector-to-target (E:T) ratio.30 Tumor cell fluorescence was measured using the Incucyte live cell imaging system as a surrogate marker of disease. 7CAR8 effectively eliminated tumor cells at all 3 E:T ratios compared with UTD and untreated cells (Figure 2E). We next established an animal model of T-ALL by engrafting CCRF-GFP-Luc cells in NSG mice to assess the efficacy of 7CAR8 in vivo using total flux measured via an In Vivo Imaging System (IVIS) spectrum as a surrogate marker of disease burden.38 After establishing disease for 10 days, mice were randomized into treatment groups and treated with 7CAR8 at 1 of 3 dose levels representing a total 25-fold dose range on day 11. 7CAR8 significantly reduced disease in a dose-dependent manner relative to UTD cells and/or saline (Dulbecco’s phosphate-buffered saline [DPBS]) control treatments (Figure 2F). All mice treated with UTD cells and DPBS were euthanized by day 19 due to illness resulting from high tumor burden. Mice treated with 7CAR8 lived to study endpoint on day 35, and those that received the highest dose level of 7CAR8 had no detectable disease following treatment (Figure 2G). Lot-to-lot variability in the potency and antitumor potential of 7CAR8 cells manufactured from 3 separate healthy donors was investigated in vitro and in vivo. 7CAR8 cells were stimulated in vitro with bead-conjugated CD7 protein, and after 18 hours the levels of IFN- γ were measured in the cell supernatant (supplemental Table 4). Although we observed lot-to-lot variability in the levels of secreted IFN-γ in vitro, there was no statistical difference in the antitumor activity of each of the 3 lots when adoptively transferred to mice bearing CCRF-GFP-Luc tumors (supplemental Figure 3). 7CAR8 cells are thus potent and effective against CD7+ CCRF-GFP-Luc cells in vitro and in vivo and display minimal variability in antitumor activity from lot to lot.
In vivo activity of 7CAR8 in T-ALL patient-derived xenograft models
We screened cell surface protein levels of CD7, PD1, and PD-L1 on 70 T-ALL samples from PDX models established from the Children’s Oncology Group Trial AALL1231 (#NCT02112916; clinicaltrials.gov) (Figure 3A; supplemental Figures 4 and 5). Of the 70 murine PDX T-ALL specimens screened, 69 specimens expressed uniform levels of human CD7 protein as measured by flow cytometry analysis. Details of the patients and their disease phenotype are provided in supplemental Table 6.
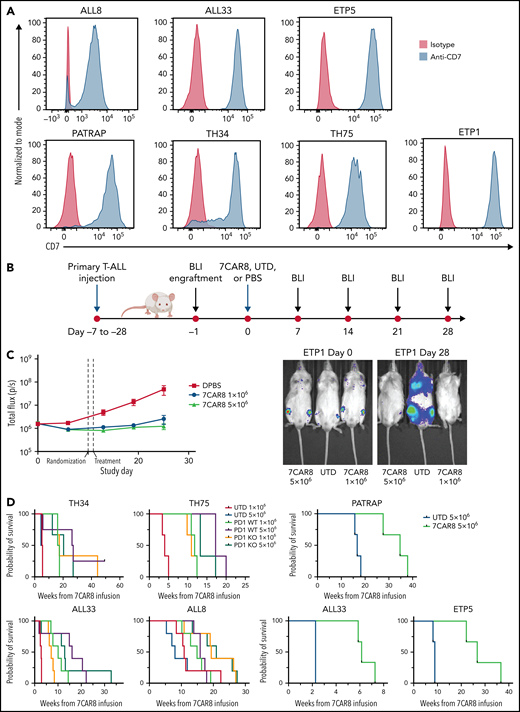
A multiplexed edited CART targeting CD7 is highly effective in improving survival in PDX of pediatric T-ALL. (A) CD7 expression on 6 unique PDX of pediatric T-ALL measured by flow cytometry are shown. Blue histograms represent CD7 expression on PDXs, and red histograms represent isotype controls. (B) Schematic of PDX T-ALL animal model studies. (C) 7CAR8 is highly effective in improving disease burden as measured by the surrogate total mean flux over time and in improving survival. A representative PDX sample of ETP T-ALL (ETP 5) was modified to express luciferase and was treated with 7CAR8. Total mean flux was significantly lower in the 7CAR8-treated arm than the UTD T-cell arm. Representative images of mice treated with UTD and 7CAR8 at day 0 and day 25 are also shown. 7CAR8 improved survival in treated mice with ETP5 (n = 3 mice per arm). (D) 7CAR8 improved survival in 5 unique PDXs. Programmed cell death protein (PD1) knock-out (KO) CART were compared with those without PD1 edit (WT) in a subset of experiments. PD1 KO CART were noninferior to PD1 WT CART in most samples. In the PDX ALL8, mice treated with the PD1 KO CART tended to survive longer than those treated with the wild type. Mice treated with either 1 × 106 or 5 × 106 UTD cells displayed no differences in survival.
A multiplexed edited CART targeting CD7 is highly effective in improving survival in PDX of pediatric T-ALL. (A) CD7 expression on 6 unique PDX of pediatric T-ALL measured by flow cytometry are shown. Blue histograms represent CD7 expression on PDXs, and red histograms represent isotype controls. (B) Schematic of PDX T-ALL animal model studies. (C) 7CAR8 is highly effective in improving disease burden as measured by the surrogate total mean flux over time and in improving survival. A representative PDX sample of ETP T-ALL (ETP 5) was modified to express luciferase and was treated with 7CAR8. Total mean flux was significantly lower in the 7CAR8-treated arm than the UTD T-cell arm. Representative images of mice treated with UTD and 7CAR8 at day 0 and day 25 are also shown. 7CAR8 improved survival in treated mice with ETP5 (n = 3 mice per arm). (D) 7CAR8 improved survival in 5 unique PDXs. Programmed cell death protein (PD1) knock-out (KO) CART were compared with those without PD1 edit (WT) in a subset of experiments. PD1 KO CART were noninferior to PD1 WT CART in most samples. In the PDX ALL8, mice treated with the PD1 KO CART tended to survive longer than those treated with the wild type. Mice treated with either 1 × 106 or 5 × 106 UTD cells displayed no differences in survival.
To evaluate the efficacy of 7CAR8 in PDX models of T-ALL, 7CAR8 cells manufactured from 3 different healthy donors were adoptively transferred into 8 different luciferase-expressing CD7+ PDX mouse models, and tumor burden was evaluated using noninvasive bioluminescent imaging (Figure 3B). 7CAR8 effectively inhibited in vivo leukemia proliferation vs UTD control treatment in all PDX models evaluated, including models of ETP and non-ETP T-ALL (representative data from PDX model ETP1 in Figure 3B; all data in supplemental Figure 6).
Survival following treatment with 7CAR8 was evaluated in 6 different PDX models. 7CAR8 significantly prolonged survival in all treated PDXs as compared with UTD T cells (Figure 3C; supplemental Tables 7 and 8). To assess the effects of PD1 KO on CART efficacy and safety, we compared 7CAR8 (PDI KO) to CART with intact PD1 expression (PD1 WT) in a subset of PDXs (TH34, TH75, ALL33, ALL8). CART with or without PD1 KO were not associated with any adverse effects in treated PDXs, and treatment with PD1 KO CARTs did not result in a significant change in survival compared with PD1 WT cells (Figure 3D). In the sample with the highest PD-L1 expression, ALL8, the PD1 KO group at the higher dose level was associated with a trend toward prolonged survival (median survival PD1 WT 5 × 106 of 15.9 weeks vs PD1 KO 5 × 106 of 21 weeks, P = .035 Mantel-Cox test; supplemental Figure 6). Thus, we concluded that 7CAR8 significantly extended the survival of mice in 6 different PDX models in a manner agnostic to PD1 editing.
Discussion
Although allogeneic CARTs derived from healthy donor T cells offer many potential advantages compared with autologous therapies, extensive modifications to enhance their clinical suitability are required. CBE is an effective technology for creating allogeneic CART via simultaneous multiplexed base editing. The use of multiplexed base editing to reduce or eliminate unwanted on-target editing outcomes has been previously demonstrated at small scale.20,29,39 Here, we demonstrate that CBE is capable of installing multiple simultaneous edits to human T cells at very high efficiencies in a clinically relevant, GMP-compliant process. We report robust proof-of-concept data for in vitro and in vivo efficacy of 7CAR8, a novel quadruple-edited CD7-targeting CART for the treatment of T-ALL. In contrast to spCas9, multiplexed editing using CBE does not cause impaired T-cell yield or activation of pathways involved in apoptosis or DNA damage response. These findings are particularly important for the development of allogeneic CART where the manufacturability and efficacy of the final therapy depends on the proliferative potential of substrate T cells.40 Furthermore, we show that unlike spCas9 editing strategies, CBE is not associated with the development of genomic rearrangement products. Thus, CBE may offer a potential pathway to minimize the unintended on-target effects of genome editing in cellular products that may be used in patients. Although 7CAR8 cells produced in 3 separate lots demonstrated no measurable cytokine-independent growth, a full assessment of the potential off-target CBE editing is important prior to clinical use.
7CAR8 was highly efficacious in vitro and in vivo in a cell line model and in multiple PDX models of T-ALL. R/r T-ALL is typically highly refractory to cytotoxic chemotherapy, and patient outcomes remain poor, despite significant overall improvements in upfront survival.22 In the setting of r/r disease, chemotherapy alone tends to be ineffective, and hematopoietic stem cell transplant (HSCT) is required for curative therapy. Due to the chemotherapy-refractory nature of the disease, HSCT is frequently not achievable as HSCT for ALL requires an MRD− remission, and this state is very difficult to obtain with conventional treatments.22 Allogeneic CART such as 7CAR8 can circumvent the challenges of harvesting T cells from patients with T-ALL and mitigate the potential for fratricide, GVHD, and recipient rejection of the allogeneic CART and may enable patients to reach MRD negativity to enable potentially curative HSCT.
Although CART cells have shown impressive efficacy in controlling and curing B-cell malignancies, their short- and long-term effectiveness may be limited by CART inhibition.41-43 Clinical trials are underway combining CART cells with PD1 inhibitors (#NCT03726515; clinicaltrials.gov). An alternative approach is to silence PD1 expression on the CART cells.43 We found that silencing PD1 expression improved polyfunctionality of 7CAR8 in vitro in a donor-dependent manner and was noninferior to wild-type PD1 CART in vivo in PDX models. However, we note that immune-compromised NSG mice are a suboptimal model to examine the efficacy of PD1 silencing or blockade and that the complexity of evaluating the effects of PD1 silencing in CART cells using animal models has resulted in several groups finding a range of potential outcomes on CART cell potency.43-45 Humanized mouse models may represent one potential avenue for future investigation of the functional relevance of PD1 silencing.
7CAR8 represents a highly efficacious, potentially curative therapy for children and adults with r/r T-ALL and has the potential for use in treating other CD7+ malignancies such as T-lymphoblastic lymphoma and subsets of acute myeloid leukemia.46 More broadly, CBE is an editing technology that enables efficient creation of multiplex-edited CART that can be used “off the shelf” with greatly reduced or eliminated unintended on-target editing outcomes that could have unforeseen clinical consequences and without impacting T-cell yield. This highly adaptable editing approach can be readily applied to additional targets. Based on these results, we plan to translate 7CAR8 into the clinic for children and adults with r/r T-ALL.
Acknowledgments
This work was supported by National Institutes of Health National Cancer Institute (grants R01CA193776, R01CA264837, R03CA256550, X01HD100702-01, and UG1CA233249) (D.T.T.); Leukemia and Lymphoma Society (D.T.T.); Children’s Oncology Group (D.T.T.); Alex’s Lemonade Stand Foundation for Childhood Cancer (D.T.T.); and The Children’s Hospital of Philadelphia Frontiers Program Immune Dysregulation Team (D.T.T.). C.D. was supported by an American Society of Clinical Oncology (ASCO) Conquer Cancer Young Investigator Award and by a Canadian Institute of Health Research (CIHR) Fellowship. These studies were supported by collaborative research funding from Beam Therapeutics (S.K.T. and D.T.T.).
Authorship
Contribution: C.D., A.E., L.B., A.C., J.C., L.C., T.F., C.G., A.L., M.L., A.M., R.M., F.M., M.N., Y.-C.P., H.P., T.R., T.V., L.Y., and Y.Z. performed experiments outline in this manuscript; S.A.G., H.N., R.S., S.K.T., G.C., J.G., and D.T.T. contributed to study design; C.D., A.E., J.G., and D.T.T. wrote the manuscript; and all authors made significant intellectual contribution to this manuscript and have seen and approved of this manuscript.
Conflict-of-interest disclosure: A.E., R.M., M.N., L.C., A.M., H.P., M.L., A.L., F.M., A.C., Y.-C.P., C.G., G.C., and J.G. were employees of Beam Therapeutics when the work was conducted and are shareholders in the company. Beam Therapeutics has filed patent applications on this work. D.T.T. serves on advisory boards for Sobi and Janssen and receives research funding from NeoImmmune Tech and Beam Therapeutics. S.A.G. receives research funding from Novartis, Kite, Vertex, and Servier and provides consultancy to Novartis, Jazz pharmaceuticals, Roche, GSK, Humanigen, CBMG, Eureka, and Janssen/JnJ. S.A.G. serves on scientific advisory boards or steering committees for Novartis, Adaptimmune, TCR2, Cellectis, Juno, Vertex, Allogene, and Cabaletta. S.K.T. receives research funding from Gilead sciences and Incyte Corporation and provides consultancy for Aleta Biotherapeutics and Kura Oncology.
Correspondence: Jason Gehrke, Beam Therapeutics, 26 Landsdowne Street, 2nd Floor, Cambridge, MA 02139; e-mail: jgehrke@beamtx.com; and Giuseppe Ciaramella, Beam Therapeutics, 26 Landsdowne Street, 2nd Floor, Cambridge, MA 02139; e-mail: gciaramella@beamtx.com.
Send data sharing requests via e-mail to the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal