

Key Points
Removal of the dominant disease allele restores telomere length in an isogenic human embryonic stem cell model of TINF2-DC.
Xenotransplantation of gene-edited human hematopoietic stem cells can be used to model disease and test potential therapeutic approaches.

Abstract
Mutations in the TINF2 gene, encoding the shelterin protein TIN2, cause telomere shortening and the inherited bone marrow (BM) failure syndrome dyskeratosis congenita (DC). A lack of suitable model systems limits the mechanistic understanding of telomere shortening in the stem cells and thus hinders the development of treatment options for BM failure. Here, we endogenously introduced TIN2-DC mutations in human embryonic stem cells (hESCs) and human hematopoietic stem and progenitor cells (HSPCs) to dissect the disease mechanism and identify a gene-editing strategy that rescued the disease phenotypes. The hESCs with the T284R disease mutation exhibited the short telomere phenotype observed in DC patients. Yet, telomeres in mutant hESCs did not trigger DNA damage responses at telomeres or show exacerbated telomere shortening when differentiated into telomerase-negative cells. Disruption of the mutant TINF2 allele by introducing a frameshift mutation in exon 2 restored telomere length in stem cells and the replicative potential of differentiated cells. Similarly, we introduced TIN2-DC disease variants in human HSPCs to assess the changes in telomere length and proliferative capacity. Lastly, we showed that editing at exon 2 of TINF2 that restored telomere length in hESCs could be generated in TINF2-DC patient HSPCs. Our study demonstrates a simple genetic intervention that rescues the TIN2-DC disease phenotype in stem cells and provides a versatile platform to assess the efficacy of potential therapeutic approaches in vivo.
Introduction
The length of telomeres, which secure genomic stability by capping the ends of linear chromosomes, is tightly regulated in adult stem cells.1-3 The telomere length reserve in the stem cell population sets the replicative potential of its differentiated progeny.4,5 For this reason, abnormally short telomeres in stem cells restrict the number of cell divisions that they or their differentiated offspring can undergo, eventually resulting in stem cell depletion and tissue failure syndromes.6-8 Telomere biology disorders (TBDs) display a broad range of clinical features, age of onset, and severity, all correlated with the extent of abnormal telomere shortening. Late-onset TBDs predominantly manifest as lung fibrosis, liver cirrhosis, and myelodysplastic syndromes. Early-onset TBDs are less frequent but are characterized by more severe syndromes.9-11 One such early-onset TBD is dyskeratosis congenita (DC), a bone marrow (BM) failure predisposition syndrome characterized by a mucocutaneous triad (oral leukoplakia, nail dystrophy, and abnormal skin pigmentation) as well as other conditions driven by premature tissue aging.9-11 The leading cause of death in DC patients is BM failure, and hematopoietic stem cell transplantation is the only definitive intervention to restore hematopoiesis.
TINF2, which encodes the TIN2 protein, is mutated in 12% of patients and thereby the second most frequently altered gene in DC cases.12 TIN2 is a member of the shelterin protein complex bridging the double-strand telomere-binding proteins TRF1/TRF2 and the TPP1/POT1 heterodimer.13 These interactions implicate a complex role of TIN2 in telomere length regulation. First, TIN2 stabilizes TRF1 on telomeres, which is a negative regulator of telomere length.14 Second, TPP1, the telomerase-recruiting protein, strictly requires TIN2 for telomere elongation and maintenance.15,16TINF2-DC mutations are uniformly heterozygous and localize to a 30 amino acid coding stretch in exon 6 called the “DC cluster”. TINF2-DC mutations usually arise de novo and result in earlier disease onset, shorter telomeres, and a more severe manifestation than other heterozygous DC-causing mutations in genes such as TERT and TERC encoding for the components of the telomerase enzyme.17-20
The mechanism by which TINF2-DC mutations cause telomere shortening is unknown. Specifically, whether telomere shortening is caused by reduced telomerase action at telomeres or by degradation of telomeric DNA remains unresolved as studies using different model systems report contrasting results.16,21 This discrepancy could be due to differences in the model systems used in the studies, highlighting the need to understand the genetic basis of the disease in a primary, preclinically relevant human model system. Previous in vitro studies of patient mutations identified in the clinic have shed light on the mode of action of a subset of TBD mutations.7,22,23 Pluripotent stem cell models with DC variants or mutations in shelterin proteins have been used to study the mechanism of aberrant telomere shortening and identify potential therapeutic avenues.24-29 Similarly, xenografts of human hematopoietic stem cells in immunocompromised mice have been used to track telomere length30 and test potential approaches to restore telomere maintenance,31 demonstrating the efficacy of such systems for studying TBDs and exploring avenues for treatments in a disease-relevant setting.
Here, we demonstrate the identification of a potential gene-targeting strategy for DC using 2 novel endogenous, isogenic model systems of TINF2-DC. Human embryonic stem cells (hESCs) engineered to express the TINF2-DC T284R mutation32,33 from the endogenous locus recapitulated the short telomere phenotype observed in DC patients. The model allowed the investigation of the mode of action of the disease mutation and the identification of a potential therapeutic gene-targeting strategy to restore telomere length and proliferative capacity in the mutant cells. Next, we used a xenotransplantation model of human hematopoietic stem and progenitor cells (HSPCs) to test the effects of our findings in more disease-relevant cells. Our work demonstrates the important aspects of the disease mechanism in vitro and the therapeutic potential of our findings in vivo.
Methods
TIN2 T284R patient mutation editing in hESCs
hESCs were electroporated with 15 μg of a CAS9 plasmid (pX33034) that contains a TINF2 exon 6 guide sequence AAAGGGAAACAGCATGACTG, 35 μg of a repair plasmid to introduce T284R mutation and a StyI restriction site, and 7.5 μg of a green fluorescent protein (GFP) expression plasmid for fluorescence-activated cell sorting. Targeting was confirmed by polymerase chain reaction (PCR) (fw: CTGAGCCCATGGAACAGAA; rev: TTGTGCCCATGGCTAGGTCT), followed by digestion with StyI.
Disruption of exon 2 of TINF2
hESCs were electroporated with 15 μg of a CAS9-P2A EGFP plasmid (pX45835) containing a TINF2 exon 2 guide sequence TCAGGACTTGGGCCCAAGGC and were sorted for GFP expression 24 hours after electroporation. Targeting of exon 2 was confirmed by PCR (fw: GTAGGACCTGGAAGGGGAAA; rev: GATCCCGCACTATAGGTCCA). Allele-specific genotyping was performed by cloning a long genomic DNA PCR product into PCR2.1 using NotI, HindIII introduced as overhangs on the PCR product with the following primers (fw: GTAGGACCTGGAAGGGGAAA; rev: GCAAGTCAACTGGGTTCTCC).
Genome editing in donor HSPCs
For genome editing of donor hCD34+ cells, CAS9 ribonucleoprotein (RNP) complex was prepared by diluting 300 pmol of in vitro-transcribed (synthesized as described36) or synthetic guide RNA (Synthego) and 500 pmol of purified CAS9 (University of California, Berkeley MacroLab) to a final volume of 20 µL of CAS9 buffer as previously described.37,38 Approximately 1 × 106 hCD34+ cells isolated from adult donor granulocyte colony-stimulating factor-mobilized peripheral blood were nucleofected using ER100 setting of Lonza 4D-Nucleofector in 100 µL of Lonza P3 solution + supplement (Lonza, V4XP-3012). Seventy-two hours after nucleofection, a fraction of the nucleofected cells were collected to assess the initial allele frequency using next-generation sequencing (NGS). The single guide RNA (sgRNA) sequences used are listed in Table 1.
sgRNA sequences used in the HSPC experiments
| sgRNA name | sgRNA sequence |
|---|---|
| TERT exon 4 | acaaucggccgcagcccguc |
| TERT exon 9 | gcuguuugcggggauucggc |
| TIN2 exon 6 | agggaaacagcaugacugug |
| TIN2 exon 2 | ucaggacuugggcccaaggc |
| sgRNA name | sgRNA sequence |
|---|---|
| TERT exon 4 | acaaucggccgcagcccguc |
| TERT exon 9 | gcuguuugcggggauucggc |
| TIN2 exon 6 | agggaaacagcaugacugug |
| TIN2 exon 2 | ucaggacuugggcccaaggc |
In vivo xenotransplantation and colony-forming unit (CFU) assay
To generate CFU colonies from hCD34+ cells 72 hours after nucleofection, a fraction of nucleofected cells was plated on methylcellulose medium (Methocult Express [StemCell Technologies, 04437])-covered 96 well plates using cell density that yields <1 colony per well. For analysis of cells after long-term in vivo xenograft, nucleofected cells were xenotransplanted in NOD.Cg-KitW-41J Tyr+ Prkdcscid Il2rgtm1Wjl/ThomJ (NBSGW) mice39 (Jackson Laboratory, 026622) via tail vein injection 72 hours after nucleofection. Eight to 16 weeks after transplantation, hCD34+ cells were sorted from the BM by fluorescence-activated cell sorting after staining with anti-human CD34 antibody (BD Biosciences, 348791) and anti-mouse CD45 antibody (BD Biosciences, 553081). A fraction of the sorted hCD34+ cells were collected for NGS, and the rest was plated on a methylcellulose medium. The resulting colonies were collected after 3 weeks in culture, genotyped, and used for single telomere length analysis (STELA).40 Mouse xenotransplantations were performed under the protocol AUP-2016-12-9389-1, approved by the office of laboratory animal care at the University of California, Berkeley. Donor and replicate information of all HSPC experiments is listed in Table 2.
Donor/replicate information of all HSPC experiments
| Replicate | Donor ID (sex) | sgRNA | Appearance in paper | Biological replicates, # (mice, #) |
|---|---|---|---|---|
| TERT exon 4 editing | ||||
| 1 | ROO3450 (male) | Synthetic | Figure 4, supplemental Figure 4 | 1 (male) |
| 2 | ROO3716 (female) | Synthetic | Figure 4, supplemental Figure 4 | 1 (male) |
| TIN2 exon 6 editing | ||||
| 1 | ROO3056 (female) | Synthetic | Figure 5, supplemental Figure 8 | 3 (1 male, 2 female) |
| 2 | ROO3056 (female) | In vitro–transcribed | 3 (1 male, 2 female) | |
| TERT exon 9 editing | ||||
| 1 | ROO3450 (male) | Synthetic | Supplemental Figure 7 | 1 (male) |
| TIN2 exon 2 editing | ||||
| 1 | ROO3446 (female) | Synthetic | Figure 6A-B, supplemental Figure 9A-C | 2 (1 male, 1 female) |
| Replicate | Donor ID (sex) | sgRNA | Appearance in paper | Biological replicates, # (mice, #) |
|---|---|---|---|---|
| TERT exon 4 editing | ||||
| 1 | ROO3450 (male) | Synthetic | Figure 4, supplemental Figure 4 | 1 (male) |
| 2 | ROO3716 (female) | Synthetic | Figure 4, supplemental Figure 4 | 1 (male) |
| TIN2 exon 6 editing | ||||
| 1 | ROO3056 (female) | Synthetic | Figure 5, supplemental Figure 8 | 3 (1 male, 2 female) |
| 2 | ROO3056 (female) | In vitro–transcribed | 3 (1 male, 2 female) | |
| TERT exon 9 editing | ||||
| 1 | ROO3450 (male) | Synthetic | Supplemental Figure 7 | 1 (male) |
| TIN2 exon 2 editing | ||||
| 1 | ROO3446 (female) | Synthetic | Figure 6A-B, supplemental Figure 9A-C | 2 (1 male, 1 female) |
Additional methods
Additional methods are described in detail in the supplement: hESC and HSPC culture, editing, fibroblast differentiation, quantitative reverse transcription PCR, immunofluorescence (IF) staining and telomere dysfunction-induced foci (TIF) analysis, telomere chromatin immunoprecipitation, telomere restriction fragment assay, immunoblotting, metaphase spread/fluorescence in situ hybridization, telomere repeat amplification protocol, human telomerase RNA component (hTERC) overexpression, telomere shortest length assay (TeSLA), STELA, and NGS analysis.
Results
Endogenous TINF2-DC mutation leads to the short telomere patient phenotype in hESCs
To assess the effect of DC-mutant TIN2 on telomere length in telomerase-positive primary cells, we introduced the TIN2-T284R mutation endogenously to recapitulate the physiological expression level of heterozygous and homozygous mutant cells (Figure 1A-B). We confirmed the presence of the mutations and their expression by sequencing the PCR-amplified genomic DNA and cDNA isolated from the mutant cells, respectively (Figure 1C; supplemental Figure 1A-C). In both the heterozygous and homozygous cell lines, TIN2 localized to telomeres as shown by colocalization with TRF1 and TPP1 (Figure 1D; supplemental Figure 1D). Telomere chromatin immunoprecipitation showed that the association of TRF1 and TPP1 with telomeres was not significantly perturbed in mutant cells (supplemental Figure 1E-F). The expression of TIN2 and other shelterin proteins in the heterozygous and homozygous cell lines was comparable to the wild-type (WT) level (supplemental Figure 1G-H). To assess telomere length, we serially passaged WT, heterozygous, and homozygous hESCs for 150 days. Throughout this time course, the average telomere lengths of heterozygous and homozygous hESCs were shorter than that of WT cells and were stable over time (Figure 1E). We demonstrated the endogenous TINF2-DC mutation led to the telomere shortening disease phenotype in hESCs.
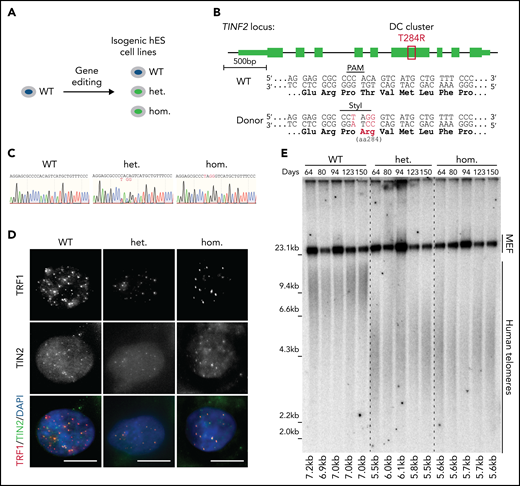
Endogenous TINF2-DC mutation leads to the short telomere patient phenotype in hESCs. (A) Experimental overview: generation of isogenic hESC lines. (B) Schematic of TINF2 locus editing by inducing CAS9-mediated double-strand break and homologous recombination in the presence of a repair template including the desired mutation. Successful targeting results in the ablation of the PAM site and de novo generation of a StyI site. (C) Sanger sequencing chromatogram of PCR amplicons of genomic DNA from the WT, heterozygous (het WT/T284R), and homozygous (hom T284R/T284R) hESC lines. (D) IF staining of telomere-binding proteins TRF1 (red) and TIN2 (green) in the WT, het, and hom hESCs. DNA was stained with 4,6-diamino-2-phenylindole (DAPI; blue). Scale bar, 10 μm. (E) Telomere length analysis of WT, het, and hom hESCs at indicated time points after targeting.
Endogenous TINF2-DC mutation leads to the short telomere patient phenotype in hESCs. (A) Experimental overview: generation of isogenic hESC lines. (B) Schematic of TINF2 locus editing by inducing CAS9-mediated double-strand break and homologous recombination in the presence of a repair template including the desired mutation. Successful targeting results in the ablation of the PAM site and de novo generation of a StyI site. (C) Sanger sequencing chromatogram of PCR amplicons of genomic DNA from the WT, heterozygous (het WT/T284R), and homozygous (hom T284R/T284R) hESC lines. (D) IF staining of telomere-binding proteins TRF1 (red) and TIN2 (green) in the WT, het, and hom hESCs. DNA was stained with 4,6-diamino-2-phenylindole (DAPI; blue). Scale bar, 10 μm. (E) Telomere length analysis of WT, het, and hom hESCs at indicated time points after targeting.
Telomere shortening occurs in the TINF2-DC stem cells and reduces the proliferative capacity of differentiated cells
The short telomeres observed in the mutant hESCs could result from excessive nucleolytic degradation caused by increased DNA damage at telomeres21 or defective telomerase recruitment.16 We did not observe increased TIFs or chromosomal abnormalities in mutant cells (Figure 2A-B; supplemental Figure 2A), suggesting that telomeres are not recognized as sites of DNA damage. We next tested whether the TINF2-DC mutation exacerbated the telomere shortening rate in telomerase-negative cells by differentiating hESCs into fibroblasts. If telomerase-independent excessive nucleolytic degradation was the cause of telomere shortening, we would expect to see increased telomere shortening rates in differentiated cells carrying the mutation. On the other hand, if TERT is epistatic to TINF2, telomere shortening rates would be similar between the mutant and WT cells. We confirmed that the levels of telomerase activity were indistinguishable between the hESC lines and undetectable after differentiation (Figure 2C). When serially passaged, fibroblasts derived from the mutant hESCs had a reduced proliferative capacity of about 10 PD compared with 25 PD of WT cells (Figure 2D). This difference can be attributed exclusively to different starting telomere lengths, not to differences in the shortening rate, which was similar in all 3 cell lines (Figure 2E; supplemental Figure 2B-C). Overexpression of the telomerase RNA component TERC in hESCs, which is limiting for telomerase activity in hESCs,41 resulted in robust telomere elongation irrespective of the TINF2 genotype (supplemental Figure 2D-E). This confirmed that telomeres in TINF2-DC mutant cells could be elongated by telomerase despite the TINF2-DC mutation. Therefore, the disease-relevant telomere shortening occurred exclusively in the telomerase-positive stem cell compartment, leading to a reduced proliferative capacity of the cell progeny.
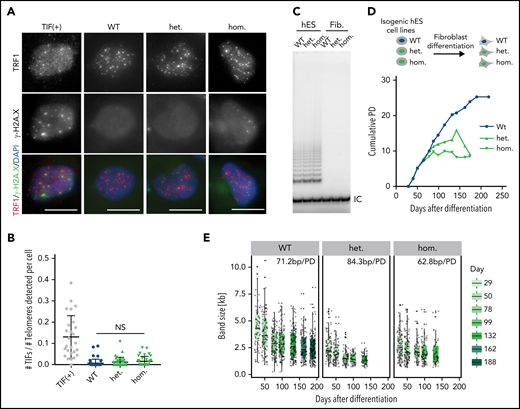
Telomere shortening occurs in the TINF2-DC stem cells and reduces the proliferative capacity of differentiated cells. (A) IF staining of TRF1 (red) and γ-H2A.X (green) of WT, heterozygous, and homozygous hESCs. TIF (+): positive control. DNA was stained with DAPI (blue). Scale bar, 10 μm. (B) Quantification of the number of TIFs detected per telomeres per cell. Mann-Whitney test. NS: P > .05. (C) Telomerase repeat amplification assay of the indicated cell lines as hESCs or fibroblasts 30 days after differentiation. IC, internal control. (D) Experimental overview of fibroblast differentiation and growth curves of cumulative population doublings (PDs) over days after differentiation. The isogenic fibroblasts lines were serially passaged until they reached replicative senescence. (E) Quantification of the size of TeSLA PCR products (n = 8) of fibroblasts at indicated time points after differentiation and telomere shortening rate in base pairs per PD (bp/PD).
Telomere shortening occurs in the TINF2-DC stem cells and reduces the proliferative capacity of differentiated cells. (A) IF staining of TRF1 (red) and γ-H2A.X (green) of WT, heterozygous, and homozygous hESCs. TIF (+): positive control. DNA was stained with DAPI (blue). Scale bar, 10 μm. (B) Quantification of the number of TIFs detected per telomeres per cell. Mann-Whitney test. NS: P > .05. (C) Telomerase repeat amplification assay of the indicated cell lines as hESCs or fibroblasts 30 days after differentiation. IC, internal control. (D) Experimental overview of fibroblast differentiation and growth curves of cumulative population doublings (PDs) over days after differentiation. The isogenic fibroblasts lines were serially passaged until they reached replicative senescence. (E) Quantification of the size of TeSLA PCR products (n = 8) of fibroblasts at indicated time points after differentiation and telomere shortening rate in base pairs per PD (bp/PD).
Telomere length and proliferative capacity can be restored by hemizygous disruption of TINF2 in hESCs
The heterozygosity of DC mutations in patients indicates that the T284R mutation is dominant. However, whether the mutant proteins are nullo/hypo, hyper, anti, or neomorphs cannot be discerned from heterozygosity alone. The clustering of DC mutations in patients to a discrete region suggests that the DC mutations impart a hyper, anti, or neomorph (gain-of-function) effect. If the DC mutation resulted in telomere shortening through a gain-of-function, disruption of the mutant allele would rescue the short telomere phenotype. If the effect of the DC mutation was by a loss-of-function mutation, disruption of the mutant allele would not affect telomere length or even might exacerbate shortening. To resolve these possibilities, we devised a strategy to remove the disease allele by introducing a single double-strand break leading to a nonhomologous end joining at exon 2 using CRISPR/CAS9 (Figure 3A). We isolated a clone with a single nucleotide insertion at the CAS9 cut site leading to a disruption of the disease allele and reduced TIN2 expression (Figure 3A-C). The clone with the hemizygous expression of WT TIN2 (WT/null) exhibited elongated telomeres and improved proliferative capacity compared with the heterozygous mutant cells (WT/T284R) (Figure 3D-E; supplemental Figure 3A) without increased chromosomal abnormalities (supplemental Figure 3B).
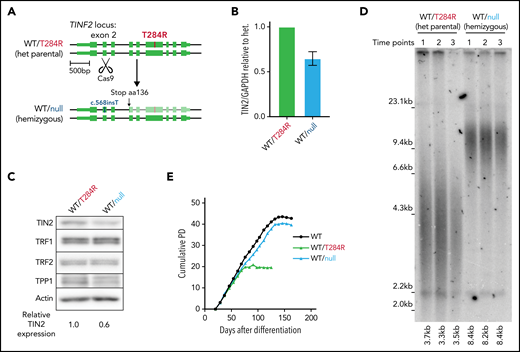
Telomere length and proliferative capacity can be restored by hemizygous disruption of TINF2 in hESCs. (A) Schematic overview: CAS9-mediated double-strand break in exon 2 to disrupt the disease allele of TINF2. (B) Relative expression level of TIN2 in the WT/null cells relative to the heterozygous (het) mutant hESCs (WT/T284R) normalized to GAPDH. Error bars indicate the SD, n = 3. (C) Western blot analysis of the het (WT/T284R) and het WT/c.568insT (WT/null) hESCs. (D) Telomere length analysis of the WT/T284R and WT/null hESCs. Each time point sample was collected weekly over 3 consecutive weeks. (E) Growth curves of cumulative PDs over days after differentiation for WT, WT/T284R, and WT/null cells.
Telomere length and proliferative capacity can be restored by hemizygous disruption of TINF2 in hESCs. (A) Schematic overview: CAS9-mediated double-strand break in exon 2 to disrupt the disease allele of TINF2. (B) Relative expression level of TIN2 in the WT/null cells relative to the heterozygous (het) mutant hESCs (WT/T284R) normalized to GAPDH. Error bars indicate the SD, n = 3. (C) Western blot analysis of the het (WT/T284R) and het WT/c.568insT (WT/null) hESCs. (D) Telomere length analysis of the WT/T284R and WT/null hESCs. Each time point sample was collected weekly over 3 consecutive weeks. (E) Growth curves of cumulative PDs over days after differentiation for WT, WT/T284R, and WT/null cells.
To generalize our findings, we generated TINF2 hemizygous cell lines by excising the region spanning exons 4 to 7 using CRISPR/CAS9 (supplemental Figure 4A-C). The disruption of TINF2 in a WT background led to telomere elongation in agreement with previous reports14 (supplemental Figure 4D, lanes 1 and 2). The removal of exons 4 to 7 in heterozygous mutant cells resulted in cell lines of 2 genotypes: hemizygous WT or hemizygous mutant (supplemental Figure 4A-C). Whether the WT or mutant allele was deleted, TINF2 hemizygous cells showed decreased expression of TIN2 (supplemental Figure 4E) and elongated telomeres compared with their parental cell line (supplemental Figure 4D). The elongated telomeres of hemizygous cells showed no signs of increased telomere DNA damage (supplemental Figure 4F-G). Our finding suggests that the disease mutation is a gain-of-function allele. Also, elongation of telomeres after TIN2 disruption without increased DNA damage and genomic instability suggests the strategy as an attractive means to restore proliferative capacity in patient cells.
Disruption of TERT in donor HSPCs demonstrates telomere shortening and proliferative disadvantage in vitro and in vivo
To assess the changes in telomere length and proliferative capacity in response to targeted gene modifications in donor HSPCs, we established a humanized mouse model. For this, we nucleofected healthy donor hCD34+ cells with CRISPR/CAS9 proteins complexed with different target sgRNAs and subjected the cells to an in vitro CFU assay or xenotransplantation in immunocompromised NBSGW mice39 (Figure 4A). To validate this system, we first disrupted the TERT locus in donor HSPCs to induce telomere shortening (supplemental Figure 5A). The most frequently occurring edited alleles were 1-nt G insertion and 8-nt deletion, leading to premature termination codons in exon 5 (supplemental Figure 5B). After editing, we assessed the telomere length and proliferative capacity of unedited and edited cells in vitro and in vivo (Figure 4A; supplemental Figure 5C). For the following experiments, we used STELA instead of TeSLA42 since STELA40 captured a wider dynamic telomere length range with less bias toward shorter telomeres (supplemental Figure 6).
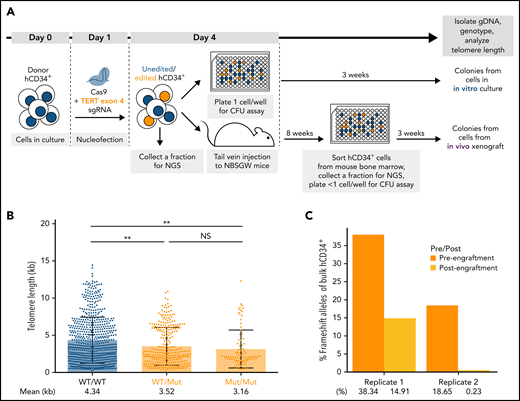
Disruption of TERT in donor HSPCs demonstrates telomere shortening and proliferative disadvantage in vitro and in vivo. (A) A schematic of the in vitro and in vivo assays to study the effect of telomere shortening caused by monoallelic or biallelic TERT disruption in donor HSPCs. (B) Telomere length analysis of CFU colonies derived from donor HSPCs 3 days after nucleofection. Mann-Whitney test. NS: P > .05 and **P < .01. (C) Percentages of frameshift alleles of bulk hCD34+ cells before and after xenotransplantation. Each replicate shows data from 1 biological replicate.
Disruption of TERT in donor HSPCs demonstrates telomere shortening and proliferative disadvantage in vitro and in vivo. (A) A schematic of the in vitro and in vivo assays to study the effect of telomere shortening caused by monoallelic or biallelic TERT disruption in donor HSPCs. (B) Telomere length analysis of CFU colonies derived from donor HSPCs 3 days after nucleofection. Mann-Whitney test. NS: P > .05 and **P < .01. (C) Percentages of frameshift alleles of bulk hCD34+ cells before and after xenotransplantation. Each replicate shows data from 1 biological replicate.
Telomere shortening was observed in the colonies with mutant alleles when cells were differentiated 3 days after editing and cultured for 3 weeks (Figure 4B). Next, we assessed the long-term effect of the mutant alleles on proliferative capacity by comparing the mutant allele frequencies of the bulk hCD34+ cells before and after xenotransplantation in mice. The mutant alleles were depreciated after xenotransplantation in both replicates (Figure 4C; supplemental Figure 5B). This suggested that the cells with disrupted TERT locus were selected against in vivo. Furthermore, we confirmed that editing with an sgRNA sequence in exon 9 led to the loss of TERT-disrupted alleles in vivo (supplemental Figure 7). Our results showed that the disruption of TERT led to telomere shortening in the short-term in vitro culture and a proliferative disadvantage in the long-term in vivo xenograft assay. Therefore, we concluded that our xenograft model could be used to assess the effects of targeted mutations on telomere length and proliferative capacity both in vitro and in vivo.
TINF2-DC alleles lead to telomere shortening but not a proliferative disadvantage in donor HSPCs in vivo
Having benchmarked the experimental system by targeting TERT, we next investigated the effect of the TINF2-DC variants in donor HSPCs (Figure 5A). We nucleofected hCD34+ cells with CAS9 RNP with sgRNA designed to cut within the DC cluster in exon 6 of the TINF2 locus. We performed the experiment in 2 replicates, 1 with CAS9 RNP only to introduce editing via nonhomologous end joining and 1 with the CAS9 RNP together with a repair template to introduce the T284R mutation via homology-directed repair (supplemental Figure 8). Since both missense and nonsense exon 6 mutations have been identified in TINF2-DC patients,17,18,43 we expected that both missense and nonsense alleles generated in this experiment would result in DC-like telomere and proliferative phenotypes. Consistent with our hESC experiments, the colonies derived from cells with mutant alleles of TINF2 exhibited shorter telomeres after xenotransplantation (Figure 5B). However, the mutant alleles did not decline in frequency throughout the xenograft experiment (Figure 5C). Thus, pathogenic indels in the DC cluster led to telomere shortening but not to an immediate proliferative disadvantage within the timeframe of our long-term in vivo assay.
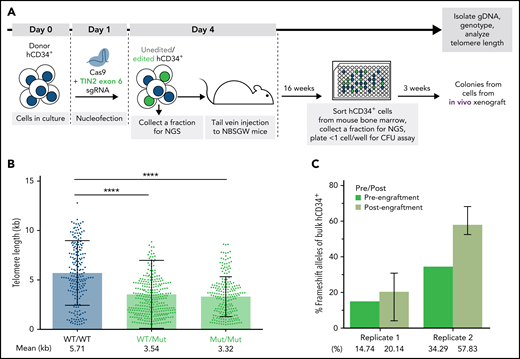
TINF2-DC alleles lead to telomere shortening but not a proliferative disadvantage in donor HSPCs in vivo. (A) A schematic of the in vivo humanized mouse model to study the effect of TINF2-DC variants in donor HSPCs. (B) Telomere length analysis of CFU colonies plotted by genotypes. Mann-Whitney test. ****P < .0001. (C) Percentages of frameshift alleles of bulk hCD34+ cells before and after engraftment in NBSGW mice. Error bars indicate data point ranges of 3 biological replicates.
TINF2-DC alleles lead to telomere shortening but not a proliferative disadvantage in donor HSPCs in vivo. (A) A schematic of the in vivo humanized mouse model to study the effect of TINF2-DC variants in donor HSPCs. (B) Telomere length analysis of CFU colonies plotted by genotypes. Mann-Whitney test. ****P < .0001. (C) Percentages of frameshift alleles of bulk hCD34+ cells before and after engraftment in NBSGW mice. Error bars indicate data point ranges of 3 biological replicates.
TINF2 disruption at exon 2 in donor and patient HSPCs
In our hESCs, telomeres shortened when we introduced a missense TINF2-DC mutation and elongated when TINF2 was disrupted upstream of the DC cluster. These observations prompted us to evaluate the disruption of TINF2 as a potential therapeutic approach and develop a protocol to disrupt TINF2 in donor HSPCs (supplemental Figure 9A). TINF2 editing in exon 2 of donor HSPCs was efficient but did not yield homozygously-edited colonies, confirming the essentiality of TINF244 (supplemental Figure 9B). In contrast to the results obtained from hESCs, the disruption of TINF2 in exon 2 in donor HSPCs did not significantly alter telomere length in vivo (Figure 6A). The relative abundance of edited alleles did not change after xenotransplantation, indicating that heterozygous TINF2 disruption did not alter the proliferative capacity of cells in vivo (Figure 6B; supplemental Figure 9C). Finally, we also introduced the TINF2 disruption in TINF2-mutant patient cells with very short telomeres (Figure 6C). We isolated ∼285 000 CD34+ cells from the patient marrow (supplemental Figure 9D) and nucleofected the cells with CAS9-RNP targeting exon 2. Five days after nucleofection, the editing efficiency of the bulk cells was 38.43% (Figure 6D). This showed the feasibility of TINF2 disruption in the patient HSPCs. Due to the limited number of cells available, changes in cellular viability or telomere length in response to the TINF2 disruption could not be assessed in these proof-of-concept editing experiments, and further assessment is warranted.
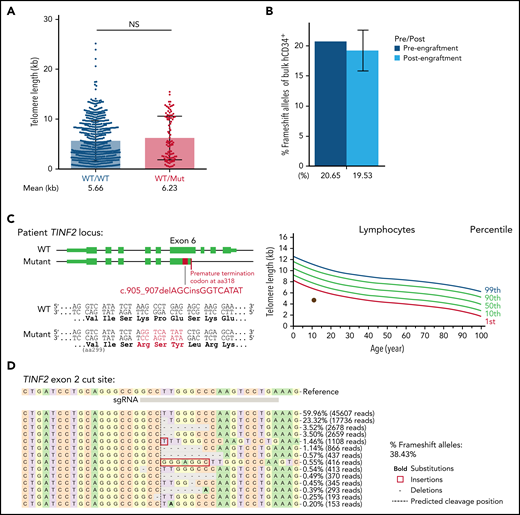
TINF2 disruption at exon 2 in donor and patient HSPCs. (A) Telomere length analysis of colonies plotted by genotype at TINF2 exon 2. Mann-Whitney test. NS: P > .05. (B) Percentages of frameshift alleles of bulk hCD34+ cells before and after engraftment in NBSGW mice. Error bars indicate data point ranges of 2 biological replicates. (C) A schematic of the patient TINF2 locus with c.905_907delAGCinsGGTCATAT mutation (left). Lymphocyte telomere length (kb) according to age in the patient (right, brown dot). (D) Allele distributions of the patient CD34+ cells 5 days after nucleofection.
TINF2 disruption at exon 2 in donor and patient HSPCs. (A) Telomere length analysis of colonies plotted by genotype at TINF2 exon 2. Mann-Whitney test. NS: P > .05. (B) Percentages of frameshift alleles of bulk hCD34+ cells before and after engraftment in NBSGW mice. Error bars indicate data point ranges of 2 biological replicates. (C) A schematic of the patient TINF2 locus with c.905_907delAGCinsGGTCATAT mutation (left). Lymphocyte telomere length (kb) according to age in the patient (right, brown dot). (D) Allele distributions of the patient CD34+ cells 5 days after nucleofection.
Discussion
By gene editing the TINF2-DC cluster region in hESCs and human HSPCs, we compared the effect of endogenous disease mutations in both telomerase-positive stem cells and isogenic, telomerase-negative differentiated cells. Using hESCs, we found that the telomere shortening caused by the TINF2-DC mutations was telomerase pathway-dependent and affected telomere length dynamics only in telomerase-positive stem cells. This was consistent with a previous finding that TINF2-DC mutations lead to short telomeres due to impaired telomerase recruitment.16 In this study, hESCs with the TINF2-DC mutations established a new telomere length set point without a growth defect, TIFs, or compromised proliferative capacity. Yet, differentiated TINF2-DC cells had a reduced proliferative capacity due to the lower telomere length set point. The replicative potential of mutant cells could be restored, together with telomere length, by disrupting 1 allele of TINF2. By employing a humanized mouse model, we translated our findings in hESCs to donor HSPCs. After demonstrating that telomerase activity is present and necessary to maintain telomere length and proliferative capacity of donor HSPCs in vivo using a TERT knockout strategy, we showed that the TINF2-DC mutations caused telomere shortening in HSPCs in vivo. However, the mutations did not lead to a marked decrease in the proliferative capacity of the hCD34+ stem and progenitor cell pool or a decrease in differentiated progenies, which was unexpected. The HSPC model also demonstrated that a loss of function of 1 allele of TINF2 did not lead to a significant increase in telomere length or proliferative capacity as it did in the hESC model.
In this study, both heterozygous and homozygous TINF2-mutant hESCs were viable in the long term and had similarly short but stable and protected telomeres. This indicates that the TINF2-DC mutation does not act by suppressing the function of the remaining WT TIN2 protein in heterozygous cells. This prompted us to investigate the consequences of excising the mutant allele. Hemizygous loss of TINF2 restored telomere length and proliferative capacity in the TINF2-DC mutant cells without inducing DNA damage responses at telomeres. This suggested that telomere regulation by TIN2 is dosage-dependent, and the disease allele acted as a dominant allele. From this, we hypothesized that disruption of the disease allele could be used to elongate telomeres and restore proliferative capacity in patient cells. This hypothesis is supported by a report of a patient with the TIN2-T284R mutation and idiopathic pulmonary fibrosis.45 The patient had the T284R mutation in the lung-derived DNA and an additional 15 base pair deletion upstream of the T284R allele in 60% of the blood-derived DNA.45 The fact that the patient did not exhibit BM failure even though they carried a TINF2 mutation suggested that the blood cells with the 15 base pair deletion upstream of the disease allele had a selective advantage and went through a clonal expansion. Since our experiments in the hESCs and the report focus specifically on the T284R mutation, further investigation is necessary to confirm if our hypothesis is applicable to other TIN2 disease variants. Nevertheless, the case reported in this study demonstrated the feasibility of our hypothesis and prompted us to evaluate the consequences of a similar disruption of the mutant TINF2-DC allele in disease-relevant HSPCs.
Xenotransplantation of polyclonal hCD34+ HSPCs without selecting for edited cells allowed us to directly compare the unedited WT and isogenic mutant cells, making our experiments internally controlled. In our in vivo TINF2-DC model, cells carrying mutant alleles had shorter telomeres but were not depleted from the stem cell pool after 16 weeks of engraftment. Moreover, the TINF2-mutant stem cells could generate differentiated progenies in vitro after being isolated from long-term engraftment. This suggested that the TINF2-DC mutant CD34+ cells retained normal engraftment and differentiation potential. Similar observations were made by Nagpal and colleagues,31 in which normal hCD34+ cells with ablated TERC did not demonstrate long-term engraftment or differentiation potential defects. The authors attributed this to an insufficient telomeric sequence loss when starting with normal donor HSPCs. On the contrary, it has been reported that TERC-DC patient cells with already short telomeres showed proliferation defects after 6 weeks in in vitro culture.46 From these observations, we reasoned that our system demonstrated the early disease progression in normal donor HSPCs starting from the acquisition of the TINF2-DC mutation. The in vivo phenotypes of our TINF2-DC mutant HSPCs and the TERC-ablated HSPCs in Nagpal and colleagues31 provide a striking contrast to what we observed in the HSPCs with hemizygous or complete TERT knockout. This was specifically unexpected because hESCs hemizygous for TERT do not show a reduction of proliferative capacity and differentiation potential.47 These differences could be because of the starting telomere length of the donor HSPCs or an unknown layer of telomere length regulation in human HSPCs. To conclusively compare the severity and differential phenotypes, the generation of TBD mutations in the same donor cells is required to ensure the same starting telomere length. The genome-editing strategy established here will provide a platform for this analysis.
Previous work by others27,28,31 and our study highlight specific therapeutic avenues for telomeropathies either by restoring telomere length in stem cells with abnormal telomere length or preventing abnormal telomere attrition in the stem cell compartment before disease progression. Our experiments demonstrated that the excision of the TINF2-DC alleles restored telomere length in DC-mutant hESCs. This reveals an attractive intervention strategy to restore proliferative capacity in TINF2-DC patient stem cells. Recently, compensatory somatic mutations in TBD patients were shown to promote clonal expansion in the hematopoietic system.48-50 These compensatory mutations include noncoding TERT promoter mutations and POT1 coding mutations as well as a cis somatic mutation on an RPA1 disease allele. Even though the patients with the compensatory mutations did not exhibit telomere lengthening effects, the mutations conferred the cells with a competitive advantage under the selective pressures of the short telomere environment.48 This demonstrates the feasibility of our approach as long as the cells with TINF2 excision exhibit clonal expansion in vivo under selective pressure, which warrants further validation.
Heterozygous loss-of-function truncation mutations in exon 5 of TINF2 have been associated with cancers,51,52 which could raise concerns against our TINF2 excision approach. However, the clonal expansion cases of somatic reversion mutations in TINF2 patients were mostly found in patients who had not developed myelodysplastic syndrome or acute myeloid leukemia.41,42 Moreover, the reversion mutations and genomic instability caused by crisis from short telomeres occurred in a mutually exclusive way in the TBD patients,48 suggesting that the compensatory mutations led to a protective effect without promoting malignancy. These reports suggest that our TINF2 excision strategy can result in clonal expansion of the cells without promoting cancer. Following the fate of cells with these somatic reversions would be highly informative in assessing the risks associated with our editing strategy. In conclusion, we expect that the genetic approaches and cell systems developed in this study will provide a starting point for further dissection of the mechanism of DC and the development of effective strategies to restore telomere function in DC patients.
Acknowledgments
The authors thank Won-Tae Kim and Alejandro Hernandez for advice during the early stages of the project, Shirin Jenkins and Emma J. van Grinsven for technical advice with experiments, and Netravathi Krishnappa for technical support with NGS. The authors also thank Titia de Lange for generously providing the #864 TIN2 antibody.
D.H. is a Chen Zuckerberg Biohub Investigator and supported by a Research Scholar Grants from the American Cancer Society (133396-RSG-19-029-01-DMC). D.H. is a Pew-Stewart Scholar for Cancer Research supported by the Pew Charitable Trusts and the Alexander and Margaret Stewart Trust. The work in the Hockemeyer laboratory was supported by the Siebel Stem Cell Institute, National Institutes of Health (NIH) R01-CA196884, the Chen Zuckerberg Biohub, and D.O.D. (W81XWH-19-1-0586). S.C. was supported by Hellman Graduate Fellowship. The work in the Bertuch Laboratory was supported by NIH R01 HL131744. The University of California, Berkeley and Boylor have filed a provisional patent application based on the data presented in the paper.
Authorship
Contribution: S.C., F.K.L., and D.H. designed the experiments with the advice of A.A.B.; S.G.R. generated the TINF2-edited hESCs; S.C. and F.K.L. performed all experiments and analyzed the data with the help of S.B.S., S.W., and G.R.; A.A.B. provided DC patient samples; and S.C., F.K.L., A.A.B., and D.H. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dirk Hockemeyer, Li Ka Shing Center University of California, Berkeley, 1951 Oxford St, Berkeley, CA 94720-3370; e-mail: hockemeyer@berkeley.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal