

TO THE EDITOR:
Recent studies have revealed the presence of clonally expanded cells with somatically acquired cancer-associated mutations within normal human tissues,1-5 including in blood from healthy elderly individuals, where these identify individuals with clonal hematopoiesis (CH).3-5 Among the most prevalent CH mutations are those seen in DNMT3A, TET2, ASXL1, and TP53,3-5 implicated as initiating mutations in myeloid malignancies.6,7 CH confers increased risk for later development of myeloid malignancies.3-5 However, most CH cases never develop any malignancy, and mechanisms enhancing transformation risk and clonal advantage of CH mutations remain unclear.5 Unraveling these mechanistic aspects of CH could greatly benefit from studies in genetically modified mice. Such studies have already provided some insights, but with conflicting results.8-10 Because the relevance of mice for modeling of CH mutations, myeloid malignancies, and cancer in general has been questioned,5,9-11 it would be important to establish to what degree mutations seen in human CH also occur spontaneously and promote clonal expansion in normal-aged mice. CH mutations have yet to be described in mice screened for spontaneous oncogeneic mutations,12 potentially because of the few mice investigated and sequencing strategies with insufficient sensitivity to detect small clones12 as human CH mutations, often occur early in life, but are often first detected in aged individuals (>70 years of age) when the clones have become large enough for detection with existing methodology.5,13 The much lower number of mouse hematopoietic stem cells (HSCs)14,15 and their shorter lifespan (2 to 3 years) suggest that CH mutations would occur at a much lower rate in mice and prove more difficult to detect than in human. However, the much smaller size of the mouse and fewer HSCs could potentially enable detection of CH clones in aged mice. We screened (supplemental methods, available on the Blood Web site) for the most common CH mutations in up to 24-month-old wild-type C57BL/6j mice, the most extensively used mouse strain for studies of normal and malignant hematopoiesis, including genetically modified mice with CH mutations.8-10 DNA isolated from single aged human (70 to 75 years; n = 6) or mouse (24 months; n = 6) HSC-derived cells was subjected to whole-genome sequencing, and bulk bone marrow (BM) of aged (n = 97) and transplanted (n = 88) mice to digital droplet polymerase chain reaction (ddPCR) analysis and error-corrected targeted DNA sequencing (ECTS). Similar to cultured fibroblasts,16 but not previously investigated for HSCs, we observed a significantly higher mutation rate (8.5-fold) in mouse compared with human HSCs (Figure 1A; supplemental Figure 1A). In line with previous reports,14,17 aged human HSCs contained ∼1000 mutations (Figure 1B). Although a 2-year old laboratory mouse approximates a 70-year-old human individual in relative lifespan, the increased mutation rate in mouse HSCs did not result in comparable mutational accumulation, as aged mouse HSC mutations were fivefold lower than in aged human HSCs (Figure 1B). Despite their differences in lifespan, mutations in aged mouse HSCs were distributed among similar genomic regions, dominated by the aging-associated COSMIC signature 1 featured by enrichment of C>T transitions at CpG dinucleotides18 in both aged human and mouse HSCs14,17 (Figure 1C-E; supplemental Figure 1B-C). Together with the estimated HSC pool size in mice vs humans,14,15 this suggests that although mutations targeted to CH-associated genes would be much more frequent in aged humans, they should also occur in most if not all aged mice (supplemental Figure 1D) and could therefore potentially be detected if promoting sufficient clonal expansion.
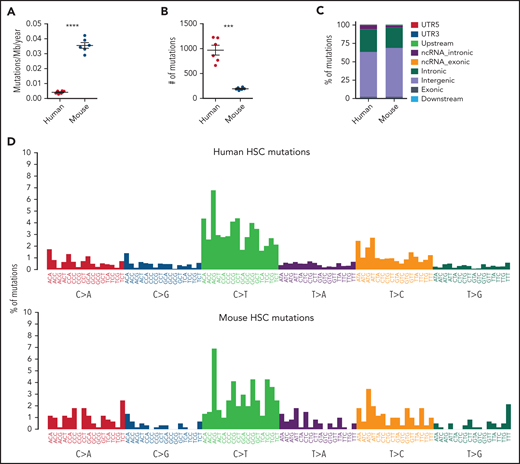

Somatic mutations in single HSCs of healthy elderly human subjects and aged wild-type mice. (A-B) Estimated yearly mutation rate per mega base pairs (Mb) (A) and total number of somatic mutations (B) in each clonally expanded HSC isolated from healthy elderly human subjects (70 to 75 years of age; n = 6) and aged wild-type mice (24 months of age; n = 6). Middle line and error bars indicate mean and standard error of the mean values, respectively. ***P < .001; ****P < .0001, Welch’s t test. (C-D) Genomic distribution and mutation signature of somatic mutations in aged human and mouse HSCs are shown in panel C. Nonnormalized mutational patterns in the context of trinucleotides are shown in panel D. (E). Hierarchically clustered heatmap showing the frequency of the COSMIC single-base substitution signatures (version 2) from each individual mouse and human HSC-derived colony as indicated by the blue scale. A dendrogram for the colonies is shown on the top. No statistical difference in contribution of mutation signatures was observed between aged mouse and human HSCs (Welch’s t test).
Somatic mutations in single HSCs of healthy elderly human subjects and aged wild-type mice. (A-B) Estimated yearly mutation rate per mega base pairs (Mb) (A) and total number of somatic mutations (B) in each clonally expanded HSC isolated from healthy elderly human subjects (70 to 75 years of age; n = 6) and aged wild-type mice (24 months of age; n = 6). Middle line and error bars indicate mean and standard error of the mean values, respectively. ***P < .001; ****P < .0001, Welch’s t test. (C-D) Genomic distribution and mutation signature of somatic mutations in aged human and mouse HSCs are shown in panel C. Nonnormalized mutational patterns in the context of trinucleotides are shown in panel D. (E). Hierarchically clustered heatmap showing the frequency of the COSMIC single-base substitution signatures (version 2) from each individual mouse and human HSC-derived colony as indicated by the blue scale. A dendrogram for the colonies is shown on the top. No statistical difference in contribution of mutation signatures was observed between aged mouse and human HSCs (Welch’s t test).
We next used highly sensitive ddPCR to screen the BM of old mice (n = 97; 24 months of age), including cells exposed to additional aging (mean, 10 months) and increased proliferation following transplantation, without any indication of hematological malignancies in any mice, for clones with recurrent human CH hotspot mutations in conserved DNA regions between mouse and human, including Jak2 V617F, Dnmt3a R878P/H/S/C/L/G (mouse equivalent of DNMT3A R882), Sf3b1 K700E/N, and Sf3b1 K666E/R/T/M/N/Q, representing 20% of reported human CH cases.3,4,19 This analysis failed to detect clones with the investigated hotspot mutations with set detection limit of 0.05% variant allele frequency (VAF) (supplemental Figure 2).
To extend the screen for CH mutations in aged mice, ECTS of coding regions in CH-associated genes (Dnmt3a, Tet2, Asxl1, Trp53, Sf3b1, Jak2, and Srsf2) was performed, capturing 82% of reported human CH mutations,3,4,19 with a detection limit <0.1% VAF (supplemental Figure 3; supplemental Tables 1 and 2). Whereas hematopoietic clones with nonsynonymous mutations in CH genes were only detected in 2% of mice at 24 months, 18% of the transplanted mice (supplemental Table 2), representing 25% of the donors used for transplantation (BM cells from each donor was transplanted into 2 recipients), showed detectable CH clones 10 months posttransplantation (Figure 2A). The 15 detected mutations were targeted to critical domains (Figure 2B-C), thereby representing likely driver mutations as in human CH. All the CH mutation-positive mice had only 1 to 2 detectable CH mutations, and identical CH mutations identified in >1 mouse were always in recipients from the same donor. ddPCR was specifically performed on sorted donor-derived cells following transplantation to ensure that mutations detected by ECTS were from aged BM donors rather than irradiated recipients (supplemental Table 3). Mutations in 2 cases, 1 Tet2 and 1 Asxl1, were confirmed in aged BM at steady state (Figure 2D), and the Tet2 mutation was also detected in both transplanted mice. In 3 cases, we detected clones with mutations in the Trp53 DNA binding domain (Figure 2E-F), including Trp53 R270H, equivalent to the TP53 R273H human cancer hotspot mutation.20 In 2 Trp53-mutated, 1 Asxl1-mutated, 1 Tet2-mutated, and 1 Dnmt3a-mutated cases (Figure 2E-F), the mutations were observed in both transplant recipients, demonstrating that these CH mutations had occurred pretransplantation but only expanded sufficiently to become detectable posttransplantation. Five CH mutations were only detected in 1 transplant recipient of aged BM (Figure 2G) and may therefore have been acquired in the aged BM posttransplantation. For 3 mutations detected posttransplantation, it was not possible to predict acquisition pre- or posttransplantation, as these were not detected in the aged BM, and the second recipient was lost during the experiment (Figure 2H). In total, we could confirm that 7 out of the 15 nonsynonymous mutations were acquired in steady state/pretransplantation and therefore not as a consequence of transplantation.

Spontaneous CH mutations in aged mice. (A) Frequency of aged (24 months) mice (n = 97) with CH mutations detected by ECTS in BM, or a mean of 10 months following transplantation into 2 lethally irradiated recipients (n = 48 donors). Mutations were only included if found to have an origin in the transplanted (CD45.2) rather than recipient (CD45.1) BM cells. ****P < .0001 Fisher’s exact test. (B-C) Distribution and characteristics of mutations detected by ECTS in BM of aged mice. Each circle in panel C represents 1 detected mutation, and the color indicates the type of mutation as indicated by the legend in panel B. (D-H) % VAF, as determined by ddPCR, of Tet2 and Asxl1 mutations detected by ECTS in aged steady-state BM (D), 2 Trp53 mutations, and 1 Asxl1 mutation in CD45.2 BM cells from 3 different donors in both recipients posttransplantation but not pretransplantation (E), Tet2 and Dnmt3a mutations with high VAF in 1 recipient and borderline VAF in both original aged donor and second recipient (F), mutations in CD45.2 BM from 1 recipient posttransplantation but not pretransplantation (G), and mutations detected only in 1 recipient posttransplant where a matched second recipient was missing (H). CD45.2 BM MNCs cells were sorted from the transplanted recipient mice. Error bars indicate 95% confidence interval, and each bar from posttransplanted mice indicates the individual recipients. #, not detected. ‡ indicates cases where ddPCR did not generate sufficient events to support confident detection (supplemental Methods).
Spontaneous CH mutations in aged mice. (A) Frequency of aged (24 months) mice (n = 97) with CH mutations detected by ECTS in BM, or a mean of 10 months following transplantation into 2 lethally irradiated recipients (n = 48 donors). Mutations were only included if found to have an origin in the transplanted (CD45.2) rather than recipient (CD45.1) BM cells. ****P < .0001 Fisher’s exact test. (B-C) Distribution and characteristics of mutations detected by ECTS in BM of aged mice. Each circle in panel C represents 1 detected mutation, and the color indicates the type of mutation as indicated by the legend in panel B. (D-H) % VAF, as determined by ddPCR, of Tet2 and Asxl1 mutations detected by ECTS in aged steady-state BM (D), 2 Trp53 mutations, and 1 Asxl1 mutation in CD45.2 BM cells from 3 different donors in both recipients posttransplantation but not pretransplantation (E), Tet2 and Dnmt3a mutations with high VAF in 1 recipient and borderline VAF in both original aged donor and second recipient (F), mutations in CD45.2 BM from 1 recipient posttransplantation but not pretransplantation (G), and mutations detected only in 1 recipient posttransplant where a matched second recipient was missing (H). CD45.2 BM MNCs cells were sorted from the transplanted recipient mice. Error bars indicate 95% confidence interval, and each bar from posttransplanted mice indicates the individual recipients. #, not detected. ‡ indicates cases where ddPCR did not generate sufficient events to support confident detection (supplemental Methods).
Except for the 15 nonsynonymous mutations, only 1 (Asxl1:c.C1584G:p.A528A) synonymous mutation was detected, resulting in a marked overrepresentation of nonsynonymous mutations (statistically significant for Trp53 and Tet2) (supplemental Table 4). Together with all the Trp53 mutations being in the DNA binding domain and most clones expanding or becoming detectable only posttransplantation, this supports that these nonsynonymous CH mutations promote clonal expansion as in humans, also in agreement with recent studies demonstrating that it can take decades and/or additional challenges before human CH clones expand sufficiently to be detectable.21-23 The relevance of increased detection of mouse CH clones following transplantation is supported by the finding that also human donor-derived CH clones undergo enhanced clonal expansion following transplantation.24,25
As could be predicted, our studies show that the lifetime of normal laboratory mice is too short to allow most clones with mutations in human CH-associated genes to undergo sufficient expansion to be detected and easily studied by available methodology. Although this will complicate or preclude CH modeling in normal aged mice, our findings of the most common human CH mutations, also in aged mice, suggest that aging of genetically modified mouse models of human CH mutations should be highly relevant for modeling the impact of human CH on normal hematopoiesis and leukemic transformation. The expansion of CH clones after transplantation and detection of the same CH mutation in multiple recipients of the same donor suggest that mouse CH mutations could be acquired early in life, but as in humans, require considerable time or challenges for the clonal expansion to be detected, which could be impacted by genetic or environmental factors, to be explored in future studies.
Authorship
Contribution: P.S.W. and S.E.W.J. conceptualized the project; D.W.L.C., T.Y., S.V.C., S.O., S.E.W.J., and P.S.W. designed the experiments; D.W.L.C., T.Y., S.V.C., F.G., and M.B. performed experiments; all authors analyzed experiments; D.W.L.C., T.Y., S.E.W.J., and P.S.W. wrote the manuscript; and all authors examined and had the opportunity to edit the manuscript and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sten Eirik W. Jacobsen, Center for Hematology and Regenerative Medicine, Karolinska Institutet, Stockholm, 141 86, Sweden; e-mail: sten.eirik.jacobsen@ki.se; and Petter S. Woll, Center for Hematology and Regenerative Medicine, Karolinska Institutet, Stockholm, 141 86, Sweden; e-mail: petter.woll@ki.se.
Targeted DNA sequencing data have been uploaded to NCBI Sequence Read Archive with accession number PRJNA767775.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal