

TO THE EDITOR:
With great interest we read a recent publication in Blood by Murali et al demonstrating somatic mutations in the MAPK/extracellular signal-regulated kinase (ERK) pathway in 60% of patients with chronic lymphocytic leukemia (CLL) with primary resistance to PI3K inhibitors.1 Different trials have underlined the efficacy of the PI3Kδ inhibitor idelalisib in combination with CD20 antibodies, such as rituximab, and charted a safety profile superior to therapies containing conventional chemotherapeutic drugs.2-4 Furthermore, next-generation PI3K inhibitors, such as copanlisib or umbralisib, promise high efficacy in lymphoid malignancies.5,6 Therefore, the understanding of resistance to PI3K inhibitors remains an important task to investigate. Furthermore, there may be differences between primary resistance with refractoriness at treatment initiation and secondary resistance, which is acquired during several months of treatment after initial response to therapy.
To this purpose, we selected patients from the CLL phase 3 trials GS-US-312-0119 (idelalisib + ofatumumab), GS-US-312-0116, and GS-US-312-0117 (both idelalisib plus rituximab)2,3 with disease progression on idelalisib. In a total of 34 patients, we performed whole-exome sequencing (WES) of tumor samples before initiation of idelalisib treatment and at the timepoint of refractory disease to identify secondary mutations acquired during therapy. In addition, in order to identify variants predicting primary refractoriness to treatment, we also assessed somatic mutations present at baseline through either WES of paired tumor and normal samples (n = 12) or targeted next-generation sequencing of up to 28 candidate genes in cases without normal control (n = 22; supplemental Table 1, available on the Blood Web site). The study was approved by local ethical review committees, and all patients gave informed consent according to the Helsinki Declaration.
The median time on idelalisib was 334 days (range, 57 to 703). Fourteen of 34 patients (41%) were nonresponders with primarily refractory (n = 2) or stable disease (SD; n = 12) as best response at a median treatment duration of 229 days (range, 57-510), whereas 19 patients initially achieved a partial remission but progressed at a median time of 506 days (range, 108-703) on treatment. All progression events occurred while patients were on the drug, and all were reviewed as CLL without Richter transformation. Measurable disease in peripheral blood and an increase of blood lymphocyte count at the time of progression were crucial criteria for analysis met by all patients as all tumor samples derived from peripheral blood.
Among the 34 patients, 11 patients had del(17p) deletion (32%) and 17 carried mutations in TP53 (50%). In total, 53% of patients (18 of 34) harbored a TP53 mutation or deletion, which was slightly more than the average in the idelalisib arms of the GS-US-312-0116 and GS-US-312-0117 trials (43%) and of the GS-US-312-0119 trial (40%).2,3 The majority of patients (85%) displayed an unmutated IGHV status (83% and 79% in the full trials). Regarding CLL driver genes, at baseline, 32% of patients carried mutations in SF3B1, 21% in NOTCH1, 12% in ATM, 9% in EGR2, 4% in BIRC3, and 4% in FBXW7 (Figure 1). Neuroblastoma RAS (NRAS) was mutated in 3 patients, and BRAF was mutated in 2 additional patients with one of them also harboring a KRAS mutation. Furthermore, we identified 2 mutations in MAPK resulting in 6 of 34 patients (17.6%) carrying mutations in the MAPK/ERK signaling pathway before treatment initiation. Among these 6 patients, 1 had PD as best response, 3 had SD, 1 had partial response, and for 1 patient the status was unknown (Figure 1). Thus, only 4 of 14 primary nonresponders to idelalisib (PD+SD) showed mutations in the MAPK/ERK pathway. Three of 5 patients with progression in the first 4 months were affected by these mutations at treatment initiation. However, we could not identify nor confidently exclude any association between response to therapy or treatment duration and MAPK/ERK-associated mutations (Student t test with P = .07 and P = .09, respectively).

Patient overview and overall genomic landscape of somatic mutations at initiation of idelalisib treatment. Samples are annotated according to tumor purity (≥80% = Y, <80% = N), performed analyses, and baseline characteristics, including treatment duration, best response, and results of central laboratory genetics before treatment initiation (baseline) with idelalisib. Patients are sorted according to treatment duration. Somatic single-nucleotide variants at the time of first sampling are provided for each patient based on WES and/or targeted next-generation sequencing (NGS). Presence of ≥ 1 mutation is marked in red, and wild-type status is marked in white. M, mutated IGHV genes; PD, progressive disease; PR, partial remission; U, unmutated IGHV genes. Gray means that the gene was not covered by any technique. The bottom row provides IGF1R expression change at progression compared with baseline in samples from 8 of the patients, with arrows indicating direction of change and gray indicating that no analysis was performed.
Patient overview and overall genomic landscape of somatic mutations at initiation of idelalisib treatment. Samples are annotated according to tumor purity (≥80% = Y, <80% = N), performed analyses, and baseline characteristics, including treatment duration, best response, and results of central laboratory genetics before treatment initiation (baseline) with idelalisib. Patients are sorted according to treatment duration. Somatic single-nucleotide variants at the time of first sampling are provided for each patient based on WES and/or targeted next-generation sequencing (NGS). Presence of ≥ 1 mutation is marked in red, and wild-type status is marked in white. M, mutated IGHV genes; PD, progressive disease; PR, partial remission; U, unmutated IGHV genes. Gray means that the gene was not covered by any technique. The bottom row provides IGF1R expression change at progression compared with baseline in samples from 8 of the patients, with arrows indicating direction of change and gray indicating that no analysis was performed.
Based on tumor/normal WES analysis (n = 12) at baseline, we identified 312 potentially pathogenic variants (mean, 26; range, 2-59). However, none of the additional findings by WES represented a key player within the MAPK/ERK or B-cell receptor signaling pathway (Figure 1).
As 19 of 34 patients in our cohort had an initial response to therapy but became refractory during treatment, we expected to find acquired mutations causing resistance to idelalisib. We found a slightly higher number of somatic mutations at progression (n = 396) in comparison with baseline (n = 312) in patients with paired tumor/normal samples (Figure 2A). In the total set, we identified 690 mutations in 629 genes that were acquired or expanded by at least 10% variant allele frequency at progression. The number of acquired mutations was not associated with response to therapy, treatment duration, or prognostic factors, such as TP53 aberrations and IGHV gene mutational status, whereas the number of mutations per year on therapy was highest in the 2 cases with PD as best response (Figure 2B).
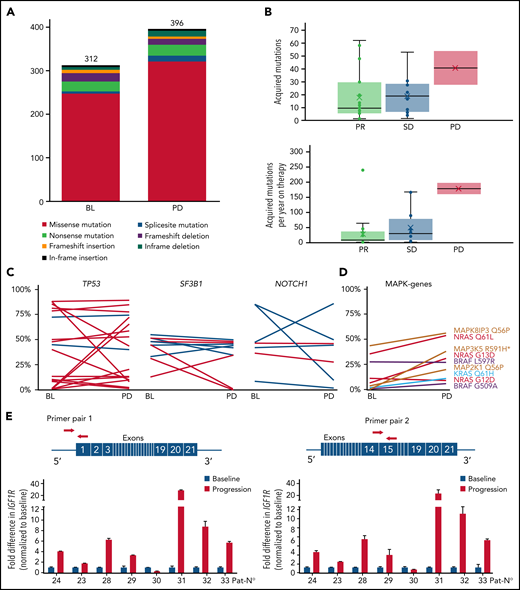
Change of gene mutations and IGF1R expression during idelalisib treatment. (A) Number of somatic mutations at baseline and at time point of progressive disease in patients with available nontumor control. (B) Acquired or expanding mutations as total number (upper panel) or per year of idelalisib treatment (lower panel) per patient in dependence of response. (C) Variant allele frequency at baseline (BL) and at timepoint of PD for single mutations in selected genes. Percentages derive from WES (dark blue) or targeted NGS (light blue) only from patients/samples with tumor cell purity of >80%. (D) Variant allele frequency of MAKP/ERK pathway mutations over time. *MAP3K5 R591H is not detected at baseline. (E) Fold difference in IGF1R expression at time of progression compared with treatment initiation calculated using ΔΔCt method with 2 different primer pairs. Red arrows denote the primer positions in IGF1R gene.
Change of gene mutations and IGF1R expression during idelalisib treatment. (A) Number of somatic mutations at baseline and at time point of progressive disease in patients with available nontumor control. (B) Acquired or expanding mutations as total number (upper panel) or per year of idelalisib treatment (lower panel) per patient in dependence of response. (C) Variant allele frequency at baseline (BL) and at timepoint of PD for single mutations in selected genes. Percentages derive from WES (dark blue) or targeted NGS (light blue) only from patients/samples with tumor cell purity of >80%. (D) Variant allele frequency of MAKP/ERK pathway mutations over time. *MAP3K5 R591H is not detected at baseline. (E) Fold difference in IGF1R expression at time of progression compared with treatment initiation calculated using ΔΔCt method with 2 different primer pairs. Red arrows denote the primer positions in IGF1R gene.
Recurrence of a mutated gene in 2 or more patients was rare and affected predominantly CLL drivers,7 such as SF3B1, NOTCH1, and TP53, which in most cases retained their clone size upon progression (Figure 2C). In addition to a newly arising MAP3K5 variant, no new mutations were identified in the MAPK/ERK pathway, whereas 4 of the 8 mutations identified at baseline expanded during therapy (Figure 2D). Other genes affecting 2 or more patients could not be assigned to a specific pathway. However, patient 1 acquired an epidermal growth factor (EGF) mutation, and patient 23 acquired a minor ERBB4 mutation, with both genes being activators of the MAPK/ERK and PI3K pathway. In addition, patient 16 acquired a TRAF2 mutation closely associated with PI3K signaling.
Taking into account all detected mutations, refractoriness did not appear to arise from major changes in the clonal composition (supplemental Figure 1). Using STRING analysis for an unsupervised network generation restricted to high-confidence associations, we found only a few of the acquired or expanded gene mutations to cluster with NRAS/MAPK and EGF (supplemental Figure 2), whereas the vast majority of patients lacked mutations with direct link to PI3K or MAPK/ERK signaling. We have recently shown in a murine model that increased IGF1R expression resulted in enhanced MAPK signaling in resistant tumors.8 In the current data set, we were able to obtain messenger RNA from treatment initiation and refractory time point in 8 patients to measure IGF1R expression. Notably, 7 of the 8 patients showed a marked upregulation of IGF1R at progression compared with baseline (Figure 2E).
Although Murali et al identified mutations within the MAPK/ERK pathway predicting primary refractoriness to idelalisib in 6 of 10 cases, we mainly focused our analysis on patients becoming refractory to idelalisib after an initial response to PI3K inhibition in analogy to recent studies on ibrutinib and venetoclax treatment.9-11 Of interest, MAPK pathway variants, although generally infrequent in CLL, were enriched in cases with primary failure to respond to idelalisib also in our data set. Among those who developed resistance to the drug after initial response, only single cases acquired genetic variants affecting MAP3K5, EGF, or ERBB4, whereas the vast majority of patients did not acquire mutations in genes that are known to directly interfere with therapy efficacy. Therefore, the addition of ERK inhibitors as proposed by Murali et al could in theory achieve responses in idelalisib nonresponders, whereas it is not so clear if it would be sufficient to control acquired refractoriness, which is much more common. However, by detecting upregulation of IGF1R, we confirmed an idelalisib resistance mechanism previously identified in the TCL1 mouse model, albeit only a subset of our patients could be analyzed. Finally, we could not identify any gatekeeper PI3Kδ mutation or mutation in any pathway that could explain acquired therapy resistance but confirmed MAPK to play a role in primary refractoriness to idelalisib.
These trials were registered at www.clinicaltrials.gov as #NCT01539512, #NCT01539291, and #NCT01659021.
Acknowledgments
This work was supported by the Else Kröner-Fresenius-Stiftung grant 2010_Kolleg24 (E.T. and S.S.), 01KT1601 (E.C.), FIRE CLL (E.C.), 031L0076C PRECISe (B.M.B.F.), Deutsche Forschungsgemeinschaft SFB 1074 projects B1, B2 (B.M.B.F.), and GILEAD (B.M.B.F.) as well as in part supported by a dedicated research grant from GILEAD (P.G.). For the Swedish center, sequencing was performed at Clinical Genomics Uppsala, SciLifeLab at Uppsala University, a national infrastructure supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. This work was supported by the Swedish Cancer Society (R.R.), the Swedish Research Council (R.R.), the Knut and Alice Wallenberg Foundation (R.R.), Karolinska Institutet (R.R.), Karolinska University Hospital (R.R.), and Radiumhemmets Forskningsfonder, Stockholm (R.R.) and by the Associazione Italiana per la Ricerca sul Cancro–AIRC, Milan, Italy Investigator Grant #20246 (P.G.).
Authorship
Contribution: E.T., S.S., P.G., R.R., and V.M. designed the research; V.M. and J.D.D. collected and analyzed the clinical data; E.T., V.L., J.D.D., M.Z., L.S., A.A., and D.Y.Y. performed genetic analyses and analyzed and interpreted the data; B.M.C.J. and A.M. measured the IGF1R expression; E.T., V.L., B.M.C.J., and D.Y.Y. performed the statistical analysis and generated figures; E.T., V.L., R.R., P.G., and S.S. wrote the first version of the manuscript; and all authors critically reviewed and approved the manuscript.
Conflict-of-interest disclosure: E.T. has received honoraria from AbbVie, Roche, and Janssen-Cilag and has received research support from AbbVie, Gilead, and Roche. R.R. has received honoraria from AbbVie, AstraZeneca, Janssen, Illumina, and Roche. P.G. has received research support from AbbVie, AstraZeneca, Gilead, Janssen, and Sunesis and has received honoraria from AbbVie, ArQule/MSD, AstraZeneca, BeiGene, Celgene/Juno/BMS, Janssen, Lilly/Loxo, Roche. V.M. was an employee of Gilead Sciences, Inc (during the time of the study) and reports stockownership of Gilead Sciences, Inc and AstraZeneca (current employment; outside the submitted work), and a family member is an employee of Gilead Sciences, Inc. S.S. has received advisory board honoraria, research support, travel support, and speaker fees from AbbVie, Amgen, AstraZeneca, Celgene, Gilead, GSK, Hoffmann-La Roche, Janssen, Novartis, and Sunesis. The remaining authors declare no competing financial interests.
Correspondence: Stephan Stilgenbauer, Division of CLL, Department of Internal Medicine III, Ulm University, Albert-Einstein-Allee 23, 89081 Ulm, Germany; e-mail: stephan.stilgenbauer@uniklinik-ulm.de.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal