

Key Points
Lung epithelial cells regulate lung inflammation during IPS through AHR suppression of AP-1 family gene expression.
A novel synthetic AHR agonist has therapeutic effects on IPS by decreasing pathogenic Th17 cells while increasing regulatory T cells.

Abstract
We previously demonstrated that interferon γ (IFN-γ) derived from donor T cells co-opts the indoleamine 2,3-dioxygenase 1 (IDO1) → aryl hydrocarbon receptor (AHR) axis to suppress idiopathic pneumonia syndrome (IPS). Here we report that the dysregulated expression of AP-1 family genes in Ahr−/− lung epithelial cells exacerbated IPS in allogeneic bone marrow transplantation settings. AHR repressed transcription of Jund by preventing STAT1 from binding to its promoter. As a consequence, decreased interleukin-6 impaired the differentiation of CD4+ T cells toward Th17 cells. IFN-γ– and IDO1-independent induction of Ahr expression indicated that the AHR agonist might be a better therapeutic target for IPS than the IDO1 activator. We developed a novel synthetic AHR agonist (referred to here as PB502) that potently inhibits Jund expression. PB502 was highly effective at inducing AHR activation and ameliorating IPS. Notably, PB502 was by far superior to the endogenous AHR ligand, L-kynurenine, in promoting the differentiation of both mouse and human FoxP3+ regulatory CD4+ T cells. Our results suggest that the IDO1-AHR axis in lung epithelial cells is associated with IPS repression. A specific AHR agonist may exhibit therapeutic activity against inflammatory and autoimmune diseases by promoting regulatory T-cell differentiation.
Introduction
Idiopathic pneumonia syndrome (IPS) is a significant noninfectious lung injury that occurs 3 to 4 months after allogeneic hematopoietic stem cell transplantation (HSCT). This disease responds poorly to therapy and has a high mortality rate.1,2 Various IPS animal models have been developed to mimic the clinical spectrum of human disease.1 In animal models, the intensity of host conditioning and donor cell alloreactivity against host tissues is important for IPS onset and its severity after HSCT.3-6 In particular, conditions causing the dysregulated activation of donor T cells result in severe IPS.5,6 Paradoxically, a lack of interferon γ (IFN-γ) signaling in host lung tissue induces severe IPS in mice through multiple mechanisms: (1) increased donor cell migration to the lung,7,8 (2) expansion of pathogenic effector T cells within the lung,7-9 (3) promotion of pathogenic Th17 cell differentiation,10,11 (4) impaired induction of programmed death ligand 1 expression,7 and (5) loss of indoleamine 2,3-dioxygenase 1 (IDO1)-mediated immune suppression.12
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor belonging to the basic helix-loop-helix/Per-Arnt-Sim homology superfamily. It is activated by various small molecules originating in the diet, microorganisms, host metabolism, and xenobiotic toxic chemicals.13,14 After binding to its ligands, AHR is released from a chaperone complex that anchors within the cytoplasm. It is translocated to the nucleus, where it functions as a transcription factor or transcriptional repressor.15 AHR was originally described as a receptor for dioxin and other xenobiotics.16 However, physiologic AHR ligands, such as tryptophan metabolites, induce the beneficial activities of AHR in hematopoietic or nonhematopoietic cells during the inflammatory response.17 For example, AHR determines the fate of immune cells by controlling the expression of cytokines, including interleukin 10 (IL-10), IL-17, IL-22, and transforming growth factor-β (TGF-β),18 and it is important for the preservation of barrier integrity and function of the gut, skin, and lung.14,19,20 IDO1 is a rate-limiting enzyme of tryptophan catabolism, and inflammatory mediators induce its expression, primarily IFN-γ, in various cell types. L-kynurenine (L-Kyn), which IDO1 synthesizes, and its breakdown products are endogenous ligands of AHR.21,22 AHR activation by these endogenous tryptophan metabolite ligands counteracts the excessive inflammatory response.23,24 Accordingly, the administration of AHR ligands effectively inhibits inflammatory tissue reactions, particularly in the lung.25-27 However, there are limitations to using AHR ligands as therapeutics because of their poor pharmacokinetics, low efficacy, and toxicity.28-30
IFN-γ derived from donor T cells induces the expression of IDO1 in lung epithelial cells and macrophages,12 and L-Kyn subsequently activates AHR during IPS. If this axis is not functional, the release of excess IL-6 by lung epithelial cells results in the differentiation of Th17 cells.11,12 AHR expression is restricted to lung epithelial cells and CD4+ T cells during IPS.12 Therefore, we hypothesized that lung epithelial cells expressing both IDO1 and AHR represent a point in which the differentiation of Th17 cells is controlled during IPS. Indeed, we show that IFN-γ and IL-1β induce the expression of IDO1 and AHR in lung epithelial cells, respectively. Consequently, activated AHR inhibits the promoter activity of Jund by blocking STAT1/JunD binding. In addition, AHR inhibits the binding of JunD to the promoter of Il6, which supports our hypothesis. Furthermore, we report a novel synthetic AHR agonist that blocks IPS concurrently by inhibiting Th17 cell differentiation and promoting FoxP3+ regulatory T-cell differentiation.
Materials and methods
Additional methods are provided in the supplemental Data, available on the Blood Web site.
Mice
Female C57BL/6 (referred to as B6.WT; H-2b), BALB/c (H-2d), and B6D2F1 (H-2b/d) mice were purchased from Charles River Laboratories. B6.Ido1−/− (H-2b), BALB/c.Ifng−/− (H-2d), and B6.Ahr+/− (H-2b) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Ahr−/− mice were bred from Ahr+/− mice, maintained in separate colonies, and used at 8 to 12 weeks of age. All animal procedures were approved by the Institutional Animal Care and Use Committee at the Inje University College of Medicine.
HSCT
B6.WT, B6.Ahr−/−, or B6.Ido1−/− mice were exposed to 2 separate doses of total-body irradiation (TBI; 850-950 cGy; cesium-137 source at 108 rad/min) within 3 hours to minimize the degree of gastrointestinal toxicity. Donor T cells were purified by AutoMACS positive selection with anti-CD90 magnetic beads (Miltenyi Biotec) from total splenocytes of BALB/c.WT and BALB/c.Ifng−/− mice. T cell–depleted (TCD) bone marrow (BM) cells were prepared from the tibia and femur of B6.WT. Through tail vein injection, the recipient mice received TCD BM cells (5 × 106) with or without purified T cells (5 × 106 wild type [WT] or 1 × 106Ifng−/−). Mice were monitored daily for survival and clinical score. The severity of graft-versus-host disease (GVHD) was assessed by the previously described scoring system, which incorporates the following 5 parameters: weight loss, posture, activity, fur texture, and skin integrity.31 Mice were scored from 0 to 2 for each criterion. A clinical index was subsequently generated by summing the 5 criterion scores (maximum index, 10).31 For BM chimerism, B6.WT or B6.Ahr−/− mice were exposed to 2 separate doses of TBI (1000 cGy). The recipient mice were administered TCD BM cells (5 × 106) of B6.WT or B6.Ahr−/− mice by tail vein injection. All recipient mice received antibiotic-containing (25 μg/mL of neomycin sulfate and 0.3 U/mL of polymyxin B sulfate) water for the first 2 weeks after HSCT. After 2 months, mixed BM chimeric mice were exposed to 2 separate doses of TBI (750 cGy). The recipient mice received transplants of TCD BM cells (5 × 106) with or without purified T cells (4 × 106) from BALB/c by tail vein injection. In the B6 → (B6 × DBA/2)F1 (BDF1; H-2b/d) acute GVHD model, lethally irradiated (950 cGy) BDF1 recipient mice received TCD BM cells (5 × 106) with or without purified T cells (4 × 106). Mice were monitored daily for survival and clinical scoring. The severity of GVHD was assessed according to the previously described scoring system.31
In vivo treatment
CH-223191 (Sigma-Aldrich) and PB502 were dissolved in Cremophor EL and ethanol (1:1) and brought to a final concentration of 1-2 mg/mL with phosphate-buffered saline. L-Kyn (Sigma-Aldrich) was dissolved in distilled water. Recipients were intraperitoneally administered 10 mg/kg of CH-223191, 100 mg/kg of L-Kyn, or 20 mg/kg of PB502 each day from day 3 through day 8 or 16 or from day 7 through day 12.
Statistical analysis
All experiments were repeated at least 2 or 3 times independently. The Mantel-Cox log-rank test was used to determine the statistical significance of the survival data. GVHD clinical scores were analyzed using a 2-way analysis of variance with the Tukey post hoc test. Normally distributed data were analyzed by a 2-tailed Student t test for single comparisons, nonparametric Mann-Whitney U test for sample sizes <6, or analysis of variance for multiple comparisons. A value of P < .05 was considered statistically significant. Data are presented as mean ± standard error of the mean.
Results
Expression of AHR is independent of the IFN-γ–IDO1 pathway in the lung after HSCT
IFN-γ derived from donor T cells induces negative feedback regulation in lung parenchyma during IPS8,10 by upregulating IDO1 expression.12 We identified several conditions for IDO1 expression in the lung,12 but how AHR expression is induced during IPS is poorly understood. Ahr expression was increased in the lung from day 3 after HSCT and reached a peak at day 5. AHR levels were maintained until day 14, from which they declined to a basal status by day 18 (Figure 1A). The expression of Cyp1b1, an AHR target gene, followed similar kinetics (Figure 1B). Recipients of TCD BM cells alone showed mildly increased expression of AHR (Figure 1C,E), indicating that upregulation of AHR expression is associated with donor T-cell responses. Immunohistochemical analysis revealed upregulated AHR expression in lung epithelial cells and donor T cells (Figure 1D). However, neither IFN-γ nor IDO1 was required for AHR expression during IPS (Figure 1E). Despite the expression of AHR in the lungs that received donor T cells, Cyp1b1 was minimally induced in the absence of Ido1 expression (Figure 1F), confirming that AHR activation of tryptophan metabolites is indispensable for Cyp1b1 expression.12 High levels of TNF-α, IL-4, and IL-13, which are secreted by inflammatory IPS-mediating Th2 cells, and IL-1β were shown to be expressed around the same time of IDO1 and AHR expression.7,12 We hypothesized these cytokines might induce AHR expression by lung epithelial cells, thereby functioning as a negative feedback regulator for IPS. Quantitative real-time polymerase chain reaction analysis showed that the combination of TNF-α and IL-1β, but not IL-4 or IL-13, effectively increased Ahr expression in isolated lung epithelial cells through NF-κB (Figure 1G). Our results suggest that Ahr expression is upregulated in lung epithelial cells under an inflammatory microenvironment regulated by NF-κB activation.
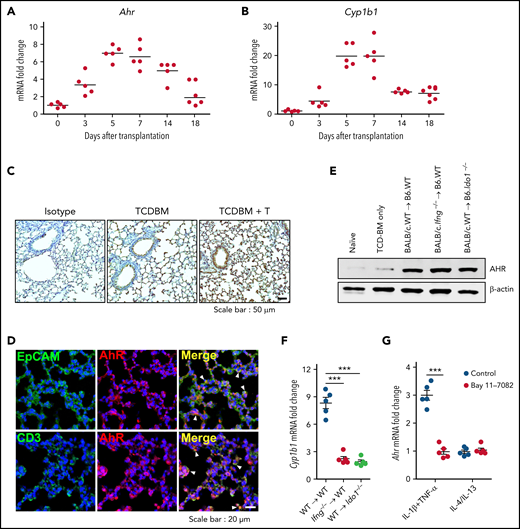
Expression of Ahr is independent of the IFN-γ–IDO1 pathway in the lung after HSCT. (A-D) Lethally irradiated B6 mice received BALB/c T cells (5 × 106) and TCD BM cells (5 × 106). Lungs were harvested at the indicated times. (A-B) Expression of Ahr (A) and Cyp1b1 (B) was measured using real-time polymerase chain reaction (PCR). Expression levels are presented as the fold change relative to the value of 1 of naïve lungs (n = 5 per time point). (C) Immunohistochemical staining of AHR on 7-day IPS lung sections. Representative images (×200) are shown. (D) Immunofluorescence staining of AHR plus EpCAM or CD3 on 7-day IPS lung sections. Blue indicates 4′,6-diamidino-2-phenylindole staining. Arrowheads indicate colocalization. Representative images (×400) are shown. (E-F) Lethally irradiated B6.WT or B6.Ido1−/− mice received BALB/c.WT (5 × 106) or BALB/c.Ifng−/− T cells (1 × 106) and BALB/c.WT TCD BM cells (5 × 106). Lungs were harvested on day 7. (E) AHR was detected by western blot analysis. (F) Levels of Cyp1b1 expression were determined by real-time PCR. Expression levels are presented as fold change relative to the value of 1 of recipient lungs of TCD BM cells only. (G) Lung epithelial cells were isolated from B6 mice and stimulated with IL-1β plus tumor necrosis factor α (TNF-α) or IL-4 plus IL-13 in the presence or absence of Bay 11-7082. After 24 hours, Ahr expression was determined by real-time PCR. Results are representative of 3 independent experiments with similar results. Data represent mean ± standard error of the mean. (F-G) One-way analysis of variance was performed. ***P < .001. mRNA, messenger RNA.
Expression of Ahr is independent of the IFN-γ–IDO1 pathway in the lung after HSCT. (A-D) Lethally irradiated B6 mice received BALB/c T cells (5 × 106) and TCD BM cells (5 × 106). Lungs were harvested at the indicated times. (A-B) Expression of Ahr (A) and Cyp1b1 (B) was measured using real-time polymerase chain reaction (PCR). Expression levels are presented as the fold change relative to the value of 1 of naïve lungs (n = 5 per time point). (C) Immunohistochemical staining of AHR on 7-day IPS lung sections. Representative images (×200) are shown. (D) Immunofluorescence staining of AHR plus EpCAM or CD3 on 7-day IPS lung sections. Blue indicates 4′,6-diamidino-2-phenylindole staining. Arrowheads indicate colocalization. Representative images (×400) are shown. (E-F) Lethally irradiated B6.WT or B6.Ido1−/− mice received BALB/c.WT (5 × 106) or BALB/c.Ifng−/− T cells (1 × 106) and BALB/c.WT TCD BM cells (5 × 106). Lungs were harvested on day 7. (E) AHR was detected by western blot analysis. (F) Levels of Cyp1b1 expression were determined by real-time PCR. Expression levels are presented as fold change relative to the value of 1 of recipient lungs of TCD BM cells only. (G) Lung epithelial cells were isolated from B6 mice and stimulated with IL-1β plus tumor necrosis factor α (TNF-α) or IL-4 plus IL-13 in the presence or absence of Bay 11-7082. After 24 hours, Ahr expression was determined by real-time PCR. Results are representative of 3 independent experiments with similar results. Data represent mean ± standard error of the mean. (F-G) One-way analysis of variance was performed. ***P < .001. mRNA, messenger RNA.
Inhibition of host AHR activation results in exacerbation of IPS
We have demonstrated that the sustained activation of STAT3 results in IDO1 expression and subsequent inhibition of IPS through AHR activation in an IFN-γ–independent fashion.12 However, it has not been determined whether donor T cell–derived IFN-γ suppresses IPS via AHR activation. Expression of Ido1 in the lung after HSCT was not affected by the AHR inhibitor CH-223191 (Figure 2A). However, administration of CH-223191 significantly downregulated Cyp1b1 expression in whole lungs, CD4+ T cells, and lung epithelial cells (Figure 2B). Cyp1b1 was expressed normally in donor CD90+ T cells of Ahr−/− recipients (Figure 2C), indicating that AHR activation in donor CD90+ T cells is not impaired in the absence of Ahr in the host. Nonetheless, deficiency of host Ahr was sufficient to induce severe IPS after HSCT concerning histopathologic scores (Figure 2D) and levels of inflammatory cytokines (Il4, Il17a, Il6, and Tnf; Figure 2E). Intracellular staining analysis showed an increase in the ratio of IL-17A+/FoxP3+ CD4+ T cells in Ahr−/− mouse lungs (Figure 2F; supplemental Figure 1A). Consistent with this, CD4+ T cells isolated from Ahr−/− recipient lungs released higher levels of IL-17A while secreting lower levels of IL-10 (Figure 2G). Most CD4+ T cells were proliferating Ki67+ cells in WT and Ahr−/− mice, but percentages of Ki67+ CD4+ T cells were greater in the latter group of mice (supplemental Figure 1C-E). It should be noted that there was a significant decrease in Il10 rather than Foxp3 expression in Ahr−/− mouse lungs (supplemental Figure 1B), indicating that AHR plays a critical role in regulating Il10 expression during IPS. Administration of CH-223191 also resulted in severe IPS in the Balb/c.WT → B6.WT model (supplemental Figure 2). To further dissect which compartment of the host hematopoietic or nonhematopoietic cells is required to develop IPS, we generated mixed BM chimeras in which either WT or Ahr−/− BM was transplanted into either WT or Ahr−/− mice. Two months after BM reconstitution, the mice were used as secondary transplant recipients. Ahr−/− recipients exhibited severe immunopathologic features of IPS, regardless of whether they were reconstituted with WT or Ahr−/− BM (Figure 2H-I). In contrast, WT mice that received either BM genotype did not develop IPS symptoms (Figure 2H-I). Our results demonstrate that Ahr deficiency in the nonhematopoietic compartment is sufficient for the development of IPS.

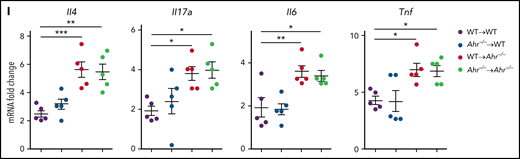
Inhibition of host AHR activation exacerbates IPS. (A-B) B6 mice that received TCD BM cells, and T cells of BALB/c mice were intraperitoneally injected with CH-223191 (10 mg/kg) every day from day 3 through day 8. Lungs were harvested on day 9. (A) Ido1, Ahr, and Cyp1b1 expression were measured by real-time polymerase chain reaction (PCR). (B) Cyp1b1 expression was measured in isolated CD4+ T cells and lung epithelial cells by real-time PCR. (C-G) B6.WT or B6.Ahr−/− mice received TCD BM cells and T cells of BALB/c mice. Lungs were harvested on day 9. (C) Cyp1b1 expression was measured in isolated CD90+ T cells and CD90− non-T cells of B6.Ahr−/− mice by real-time PCR. (D) Representative images (×200; left) and histopathologic scoring (right). (E) Levels of cytokine messenger RNA (mRNA) were measured in whole lungs by real-time PCR. (F-G) Single-cell suspensions of lung tissue were stimulated with anti-CD3/CD28 for 3 hours for fluorescence-activated cell sorting analysis or 24 hours for harvesting culture supernatant (n = 5 per group). (F) Staining of CD4 and IL-17A or FoxP3 were performed, and ratio of IL-17A+/FoxP3+ CD4+ T cells was obtained (n = 5 per group). (G) Concentrations of IL-17A and IL-10 were measured in culture supernatant. (H-I) Mixed chimeric mice were generated by transplanting B6.WT or B6.Ahr−/− BM into lethally irradiated B6.WT or B6.Ahr−/− mice. After 2 months, the chimeric mice received TCD BM cells and T cells of BALB/c mice. Lungs were harvested on day 9 (n = 5 per group). (H) Representative images (200×; left) and histopathologic scoring (right). (I) Levels of cytokine mRNA were measured by real-time PCR. Results are representative of 2 (B-C,H-I) or 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (A-G) and 1-way analysis of variance (H-I) were performed. *P < .05, **P < .01, ***P < .001.
Inhibition of host AHR activation exacerbates IPS. (A-B) B6 mice that received TCD BM cells, and T cells of BALB/c mice were intraperitoneally injected with CH-223191 (10 mg/kg) every day from day 3 through day 8. Lungs were harvested on day 9. (A) Ido1, Ahr, and Cyp1b1 expression were measured by real-time polymerase chain reaction (PCR). (B) Cyp1b1 expression was measured in isolated CD4+ T cells and lung epithelial cells by real-time PCR. (C-G) B6.WT or B6.Ahr−/− mice received TCD BM cells and T cells of BALB/c mice. Lungs were harvested on day 9. (C) Cyp1b1 expression was measured in isolated CD90+ T cells and CD90− non-T cells of B6.Ahr−/− mice by real-time PCR. (D) Representative images (×200; left) and histopathologic scoring (right). (E) Levels of cytokine messenger RNA (mRNA) were measured in whole lungs by real-time PCR. (F-G) Single-cell suspensions of lung tissue were stimulated with anti-CD3/CD28 for 3 hours for fluorescence-activated cell sorting analysis or 24 hours for harvesting culture supernatant (n = 5 per group). (F) Staining of CD4 and IL-17A or FoxP3 were performed, and ratio of IL-17A+/FoxP3+ CD4+ T cells was obtained (n = 5 per group). (G) Concentrations of IL-17A and IL-10 were measured in culture supernatant. (H-I) Mixed chimeric mice were generated by transplanting B6.WT or B6.Ahr−/− BM into lethally irradiated B6.WT or B6.Ahr−/− mice. After 2 months, the chimeric mice received TCD BM cells and T cells of BALB/c mice. Lungs were harvested on day 9 (n = 5 per group). (H) Representative images (200×; left) and histopathologic scoring (right). (I) Levels of cytokine mRNA were measured by real-time PCR. Results are representative of 2 (B-C,H-I) or 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (A-G) and 1-way analysis of variance (H-I) were performed. *P < .05, **P < .01, ***P < .001.
AHR inhibits differentiation of Th17 cells through regulation of gene expression in lung epithelial cells
Because lung epithelial cells are the major host cell type expressing AHR during IPS (Figure 1E), we hypothesized that lung epithelial cell AHR may be a key regulator for the differentiation of donor CD4+ T cells toward Th17 cells. Initially, we examined whether gene expression in lung epithelial cells creates a cytokine microenvironment favorable for Th17 differentiation in the lung of Ahr−/− recipients. Lung epithelial cells isolated from Ahr−/− recipients exhibited a gene expression pattern that largely recapitulated whole lungs, as shown in Figure 2E (Figure 3A). It was evident that they expressed higher levels of Il6 and Il1b, which are required for Th17 differentiation (Figure 3A). Stimulation of isolated lung epithelial cells with IFN-γ increased Ido1 and Cyp1b1 expression in WT mice (Figure 3B). Expression of Cyp1b1 was not upregulated in Ahr−/− lung epithelial cells by IFN-γ (Figure 3B), indicating that AHR of lung epithelial cells was sufficient to induce Cyp1b1 expression in the presence of IFN-γ under our culture conditions. A deficiency of Ahr in lung epithelial cells increased the expression of Il6 and Tnf induced by IFN-γ/IL-1β (Figure 3C-D). The addition of conditioned media from Ahr−/− lung epithelial cells stimulated with IFN-γ/IL-1β significantly increased IL-17A production by CD4+ T cells in the presence of IL-23 and TGF-β compared with WT lung epithelial cells (Figure 3E). Similarly, human peripheral CD4+ T cells produced higher levels of IL-17A in response to conditioned media from A549 cells stimulated with IFN-γ and IL-1β in the presence of CH-223191 (Figure 3F). Thus, lung epithelial cells can suppress Th17 cell differentiation through AHR during inflammatory conditions.
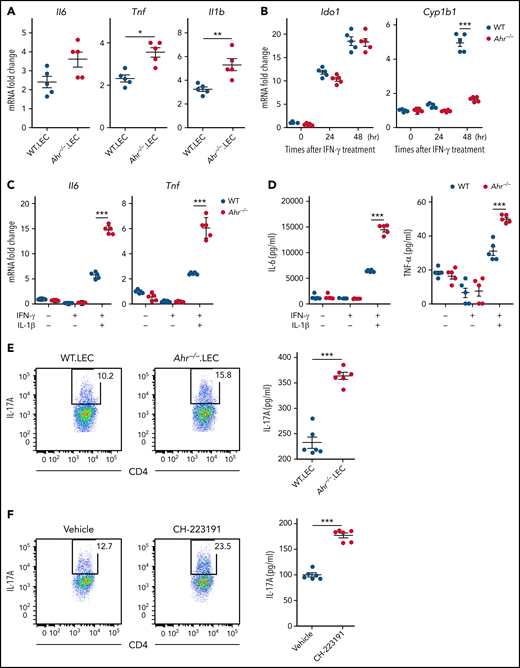
Regulation of gene expression by AHR is cell intrinsic in lung epithelial cells. (A) Lethally irradiated B6.WT or B6.Ahr−/− mice were injected with TCD BM cells plus T cells from BALB/c mice. Lung epithelial cells were isolated on day 9 (n = 5 per group). Cytokine messenger RNA (mRNA) levels were measured by real-time polymerase chain reaction (PCR). (B-D) Lung epithelial cells were isolated from naïve B6.WT or B6.Ahr−/− mice and stimulated with IFN-γ. (B) Levels of Ido1 and Cyp1b1 expression were measured by real-time PCR. (C-D) Lung epithelial cells were treated with IL-1β after 48-hour stimulation with IFN-γ. (C) Levels of cytokine mRNA were measured using real-time PCR. (D) Cytokine levels in cell culture supernatants were measured using the mouse Flex-Set cytokine bead array. (E-F) Mouse splenic (E) or human peripheral (F) CD4+ T cells were preactivated with anti-CD3/CD28 and IL-2 for 48 hours and then reactivated in the presence of combined IL-23, TGF-β, and conditioned media of WT or Ahr−/− lung epithelial cells (E) or A549 cells treated with or without CH-223191 (F). Lung epithelial cells and A549 cells were cultured as described in panel C. Representative dot plots for IL-17A+CD4+ T cells and concentrations of culture supernatant are presented. IL-17A in cell culture supernatants was measured using the mouse Flex-Set cytokine bead array. Results are representative of 2 (E-F) or 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (A), 2-way analysis of variance (B-D), and 2-tailed Student t test (E-F) were performed. *P < .05, **P < .01, ***P < 0.001.
Regulation of gene expression by AHR is cell intrinsic in lung epithelial cells. (A) Lethally irradiated B6.WT or B6.Ahr−/− mice were injected with TCD BM cells plus T cells from BALB/c mice. Lung epithelial cells were isolated on day 9 (n = 5 per group). Cytokine messenger RNA (mRNA) levels were measured by real-time polymerase chain reaction (PCR). (B-D) Lung epithelial cells were isolated from naïve B6.WT or B6.Ahr−/− mice and stimulated with IFN-γ. (B) Levels of Ido1 and Cyp1b1 expression were measured by real-time PCR. (C-D) Lung epithelial cells were treated with IL-1β after 48-hour stimulation with IFN-γ. (C) Levels of cytokine mRNA were measured using real-time PCR. (D) Cytokine levels in cell culture supernatants were measured using the mouse Flex-Set cytokine bead array. (E-F) Mouse splenic (E) or human peripheral (F) CD4+ T cells were preactivated with anti-CD3/CD28 and IL-2 for 48 hours and then reactivated in the presence of combined IL-23, TGF-β, and conditioned media of WT or Ahr−/− lung epithelial cells (E) or A549 cells treated with or without CH-223191 (F). Lung epithelial cells and A549 cells were cultured as described in panel C. Representative dot plots for IL-17A+CD4+ T cells and concentrations of culture supernatant are presented. IL-17A in cell culture supernatants was measured using the mouse Flex-Set cytokine bead array. Results are representative of 2 (E-F) or 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (A), 2-way analysis of variance (B-D), and 2-tailed Student t test (E-F) were performed. *P < .05, **P < .01, ***P < 0.001.
AHR regulates gene expression in lung epithelial cells through transcriptional repression of Jund
AHR regulates a variety of genes by interacting with other transcription factors.14,17 To investigate how AHR represses inflammatory gene expression in lung epithelial cells, we performed an RNA sequencing analysis of whole lungs from WT or Ahr−/− recipients at day 5 after HSCT. We identified an overrepresentation of the activator protein-1 (AP-1) family of transcription factors in the Ahr−/− lung (Figure 4A). Among these, Jund was markedly increased in lung epithelial cells and the whole lung from Ahr−/− recipients (Figure 4B-C). In vitro stimulation of isolated Ahr−/− lung epithelial cells with IL-1β and IFN-γ significantly increased Fos and Jund expression compared with WT lung epithelial cells (Figure 4D). In this culture system, pharmacologic inhibition of AP-1 with tanshinone IIA reduced IL-1β– and IFN-γ–induced expression of Il6 and Tnf (Figure 4E). These results are consistent with those showing AP-1 to be a key transcriptional factor that induces the expression of inflammatory cytokine genes after stimulation with inflammatory stimuli, such as IL-1β and TNF-α.32,33


AHR regulates Jund expression in lung epithelial cells. (A-C) Lethally irradiated B6.WT or B6.Ahr−/− mice received TCD BM cells and T cells from BALB/c mice. (A) Bulk RNA sequencing analysis of 5-day whole lungs. Heat map visualization of log2 (fold change; Ahr−/−/WT) is presented. (B-C) AP-1 family gene expression levels were measured in whole lungs (B) or isolated lung epithelial cells (C) by real-time polymerase chain reaction (PCR). (D) Isolated lung epithelial cells from B6.WT or B6.Ahr−/− mice were stimulated with IFN-γ for 48 hours, and IL-1β was added and further cultured for 24 hours. Jund and Fos expression levels were measured using real-time PCR. (E) Isolated lung epithelial cells from B6.WT mice were stimulated with IFN-γ in the presence or absence of tanshinone IIA (1 μM) for 48 hours, and IL-1β was added and further cultured for 24 hours. Il6 and Tnf expression levels were measured using real-time PCR. (F) A549 cells were cultured in the presence of L-Kyn (100 μM) with or without CH-223291 (10 μM) for 48 hours and further cultured in the presence of IL-1β for 24 hours. Levels of Jund and Il6 expression were measured by real-time PCR. (G) A549 cells were stimulated with IL-1β in the presence of the indicated inhibitors for 24 hours. Levels of Jund were measured by real-time PCR. (H) A549 cells were stimulated with IL-1β for 1 hour and further cultured in the presence of L-Kyn for 2 hours. Chromatin immunoprecipitation (ChIP) assays were performed to assess the binding of AHR to the xenobiotic response element site of the Jund promoter. (I-J) A549 cells were stimulated with IL-1β for 1 hour and further cultured in the presence of L-Kyn for 2 hours. ChIP assays were performed. (I) Binding of STAT1 (left) and JunD (right) to STAT1- and JunD-binding site of the Jund promoter, respectively. (J) Binding of JunD to a JunD-binding site of the Il6 promoter. Results are representative of 2 (A,I) or 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (B-E) and 1-way analysis of variance (F-J) were performed. *P < .05, **P < .01, ***P < .001. mRNA, messenger RNA.
AHR regulates Jund expression in lung epithelial cells. (A-C) Lethally irradiated B6.WT or B6.Ahr−/− mice received TCD BM cells and T cells from BALB/c mice. (A) Bulk RNA sequencing analysis of 5-day whole lungs. Heat map visualization of log2 (fold change; Ahr−/−/WT) is presented. (B-C) AP-1 family gene expression levels were measured in whole lungs (B) or isolated lung epithelial cells (C) by real-time polymerase chain reaction (PCR). (D) Isolated lung epithelial cells from B6.WT or B6.Ahr−/− mice were stimulated with IFN-γ for 48 hours, and IL-1β was added and further cultured for 24 hours. Jund and Fos expression levels were measured using real-time PCR. (E) Isolated lung epithelial cells from B6.WT mice were stimulated with IFN-γ in the presence or absence of tanshinone IIA (1 μM) for 48 hours, and IL-1β was added and further cultured for 24 hours. Il6 and Tnf expression levels were measured using real-time PCR. (F) A549 cells were cultured in the presence of L-Kyn (100 μM) with or without CH-223291 (10 μM) for 48 hours and further cultured in the presence of IL-1β for 24 hours. Levels of Jund and Il6 expression were measured by real-time PCR. (G) A549 cells were stimulated with IL-1β in the presence of the indicated inhibitors for 24 hours. Levels of Jund were measured by real-time PCR. (H) A549 cells were stimulated with IL-1β for 1 hour and further cultured in the presence of L-Kyn for 2 hours. Chromatin immunoprecipitation (ChIP) assays were performed to assess the binding of AHR to the xenobiotic response element site of the Jund promoter. (I-J) A549 cells were stimulated with IL-1β for 1 hour and further cultured in the presence of L-Kyn for 2 hours. ChIP assays were performed. (I) Binding of STAT1 (left) and JunD (right) to STAT1- and JunD-binding site of the Jund promoter, respectively. (J) Binding of JunD to a JunD-binding site of the Il6 promoter. Results are representative of 2 (A,I) or 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (B-E) and 1-way analysis of variance (F-J) were performed. *P < .05, **P < .01, ***P < .001. mRNA, messenger RNA.
To further understand how AHR represses AP-1–induced expression of inflammatory cytokines in lung epithelial cells, we examined human A549 lung epithelial cells. Treatment with IFN-γ resulted in expression of Ido1 and production of its metabolite, L-Kyn, in a time-dependent manner (supplemental Figure 3A). These cells were able to upregulate the expression of Cyp1b1 in response to L-Kyn or IFN-γ in an AHR-dependent manner (supplemental Figure 3B-C). In contrast, IFN-γ increased Il6 expression under conditions of AHR inhibition (supplemental Figure 3C). These results indicate that A549 cells are similar to primary lung epithelial cells. Importantly, L-Kyn completely inhibited IL-1β–induced expression of Jund and Il6, and this effect was completely reversed by AHR inhibition (Figure 4F). In addition, IL-1β required STAT1 and AP-1 for the expression of Jund (Figure 4G). Chromatin immunoprecipitation assays revealed that L-Kyn induced binding of AHR to its putative binding site on the Jund promoter in the absence or presence of IL-1β (Figure 4H). However, L-Kyn almost completely inhibited IL-1β–induced binding of STAT1 to the promoter of Jund, while partially inhibiting that of JunD (Figure 4I). Similarly, JunD did not bind to the promoter of Il6 when L-Kyn was administered with IL-1β (Figure 4J). These results indicate that AHR blocks the transcriptional activity of STAT1 and JunD, thereby repressing the expression of inflammatory genes in lung epithelial cells.
Therapeutic effects of AHR activation on IPS
As shown in Figure 1D, the expression of Ahr was not dependent upon IDO1 or IFN-γ. Ahr expression levels were higher in the lungs of mice that received Ifng−/− donor T cells (Figure 5A-B). Based on this observation, we evaluated the effects of the AHR ligand on IPS. We used the B6.Ifng−/−-into-Balb/c lung GVHD model, in which the recipients exhibit severe IPS. Daily injection of L-Kyn with or without CH-223191 was performed from day 3 through day 8 after HSCT. We confirmed that the expression of Cyp1b1 was upregulated in L-Kyn–injected recipients (Figure 5C). Lung histopathology clearly showed that injection of L-Kyn reduced the progression of IPS (Figure 5D). IPS amelioration by L-Kyn was accompanied by downregulation of the proinflammatory cytokines Il4, Il17a, Il6, and Tnf and upregulation of the immunosuppressive genes Il10, Tgfb1, and Entpd1 (which encodes CD39; Figure 5E-F). However, the injection of CH-223191 abolished the L-Kyn effect on IPS to a large extent (Figure 5D-F). Furthermore, Ahr−/− recipients exhibited no therapeutic effects from L-Kyn treatment, indicating that AHR activation in the host compartment is indispensable for IPS suppression (supplemental Figure 4). These results suggest that AHR activation results in the amelioration of IPS.

Therapeutic effects of L-Kyn on IPS. (A-B) Lethally irradiated B6.WT mice were injected with BALB/c.Ifng−/− T cells (1 × 106) and BALB/c.WT TCD BM cells. Lungs were harvested on day 7 (n = 5 per group). (A) Levels of Ahr expression were measured by real-time polymerase chain reaction (PCR). (B) Immunohistochemical staining of AHR. Representative images (×200) are shown. (C-F) GVHD was induced as described in panels A and B. Recipients were intraperitoneally injected with L-Kyn (100 mg/kg) with or without CH-223191 (10 mg/kg) each day from day 3 through day 8. Lungs were harvested on day 9 (C) or 14 (D-F). (C) Cyp1b1 expression was measured by real-time PCR (n = 5 per group). (D) Representative images of hematoxylin and eosin staining (×200; left) and histopathologic scoring (right; n = 5 per group). (E-F) Levels of cytokine messenger RNA (mRNA) were measured by real-time PCR (n = 5 per group). Results are representative of at least 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (A) and 1-way analysis of variance (C-F) were performed. *P < .05, **P < .01, ***P < .001.
Therapeutic effects of L-Kyn on IPS. (A-B) Lethally irradiated B6.WT mice were injected with BALB/c.Ifng−/− T cells (1 × 106) and BALB/c.WT TCD BM cells. Lungs were harvested on day 7 (n = 5 per group). (A) Levels of Ahr expression were measured by real-time polymerase chain reaction (PCR). (B) Immunohistochemical staining of AHR. Representative images (×200) are shown. (C-F) GVHD was induced as described in panels A and B. Recipients were intraperitoneally injected with L-Kyn (100 mg/kg) with or without CH-223191 (10 mg/kg) each day from day 3 through day 8. Lungs were harvested on day 9 (C) or 14 (D-F). (C) Cyp1b1 expression was measured by real-time PCR (n = 5 per group). (D) Representative images of hematoxylin and eosin staining (×200; left) and histopathologic scoring (right; n = 5 per group). (E-F) Levels of cytokine messenger RNA (mRNA) were measured by real-time PCR (n = 5 per group). Results are representative of at least 3 independent experiments with similar results. Data represent mean ± standard error of the mean. Nonparametric Mann-Whitney U test (A) and 1-way analysis of variance (C-F) were performed. *P < .05, **P < .01, ***P < .001.
Characterization of a novel synthetic AHR agonist (PB502) in lung epithelial cells
Our findings indicate that AHR is a promising target for IPS therapy. Its endogenous ligands have inherent limitations in their application to therapeutics because of poor pharmacokinetics, low efficacy, or toxicity.28-30 Therefore, we synthesized several novel AHR agonists. We screened a proprietary small-molecule library and identified several compounds that induce Cyp1a1/Cyp1b1 expression. We identified a scaffold with an indole-3-acetamide group, which was selected for further chemical optimization. Extensive structure-activity-relationship studies on the scaffold resulted in identification of PB502, a highly potent and selective AHR activator. PB502 is an indole-3-acetamide conjugated with a benzothiazole moiety (Figure 6A). Homology modeling and molecular dynamic simulation revealed that PB502 was fitted into the pocket of AHR (δ energy = −10.5 kcal/mol; Figure 6B). The T-shaped π-π interaction between His291 of AHR and the benzothiazole ring of PB502 stabilized the docking complex. In addition, the hydrogen bonds between Ser365 and Gln386 of AHR and the carbonyl oxygen of PB502 contributed to the stability of the docking complex (Figure 6B). Treatment of A549 cells with PB502 induced the nuclear translocation of AHR and subsequent binding to the xenobiotic response element of the Cyp1a1 promoter (Figure 6C-D). PB502-mediated transcription activation of Cyp1b1 and its activity were AHR dependent (Figure 6E-F). In addition, PB502 inhibited IL-1β–mediated expression of Jund and Il6 (Figure 6G). Like L-Kyn, PB502 formed an AHR-STAT1-JunD complex in response to IL-1β (Figure 6H) and blocked STAT1 and JunD binding to the Jund promoter (Figure 6I). Our results demonstrate that PB502 is a novel synthetic activator of AHR that can potently inhibit inflammatory gene expression in lung epithelial cells.

Characterization of a new synthetic AHR agonist (PB502). (A) Chemical structure of PB502. (B) Modeling of a docking site on AHR for PB502. (C) Western blot analysis of AHR in nuclear and cytoplasmic extracts of A549 cells cultured for 4 hours in the presence of PB502 (2 μM) or L-Kyn (100 μM). (D) A549 cells were cultured in the presence of PB502 or L-Kyn for 2 hours. Chromatin immunoprecipitation (ChIP) assays were performed to assess the binding of AHR to a xenobiotic response element site in the Cyp1b1 promoter. (E-F) A549 cells were cultured in the presence of PB502 with or without CH-223191 for 4 (E) or 24 (F) hours. (E) Levels of Cyp1b1 expression were measured by real-time polymerase chain reaction (PCR). (F) The enzymatic activity of CYP1A1 was measured using EROD assays. (G) A549 cells were cultured in the presence of PB502 (1 μM) with or without CH-223291 (10 μM) for 48 hours, and IL-1β was added and further cultured for 24 hours. Levels of Jund and Il6 expression were measured by real-time PCR. (H-I) A549 cells were stimulated with IL-1β for 1 hour, and PB502 or L-Kyn was added for 2 hours. Immunoprecipitation with anti-AHR antibody was performed in cell lysates, and AHR, STAT1, and JunD were detected by western blot analysis. (I) ChIP assays were performed to assess binding of STAT1 (left) and JunD (right) to the Jund promoter. Results are representative of 2 (H-I) or ≥ 4 (C-G) independent experiments with similar results. Data represent mean ± SEM. One-way analysis of variance was performed (D-G,I). *P < .05, **P < .01, ***P < .001. mRNA, messenger RNA.
Characterization of a new synthetic AHR agonist (PB502). (A) Chemical structure of PB502. (B) Modeling of a docking site on AHR for PB502. (C) Western blot analysis of AHR in nuclear and cytoplasmic extracts of A549 cells cultured for 4 hours in the presence of PB502 (2 μM) or L-Kyn (100 μM). (D) A549 cells were cultured in the presence of PB502 or L-Kyn for 2 hours. Chromatin immunoprecipitation (ChIP) assays were performed to assess the binding of AHR to a xenobiotic response element site in the Cyp1b1 promoter. (E-F) A549 cells were cultured in the presence of PB502 with or without CH-223191 for 4 (E) or 24 (F) hours. (E) Levels of Cyp1b1 expression were measured by real-time polymerase chain reaction (PCR). (F) The enzymatic activity of CYP1A1 was measured using EROD assays. (G) A549 cells were cultured in the presence of PB502 (1 μM) with or without CH-223291 (10 μM) for 48 hours, and IL-1β was added and further cultured for 24 hours. Levels of Jund and Il6 expression were measured by real-time PCR. (H-I) A549 cells were stimulated with IL-1β for 1 hour, and PB502 or L-Kyn was added for 2 hours. Immunoprecipitation with anti-AHR antibody was performed in cell lysates, and AHR, STAT1, and JunD were detected by western blot analysis. (I) ChIP assays were performed to assess binding of STAT1 (left) and JunD (right) to the Jund promoter. Results are representative of 2 (H-I) or ≥ 4 (C-G) independent experiments with similar results. Data represent mean ± SEM. One-way analysis of variance was performed (D-G,I). *P < .05, **P < .01, ***P < .001. mRNA, messenger RNA.
Therapeutic effects of PB502 on IPS
We investigated the therapeutic effect of PB502 on IPS using L-Kyn as a reference. PB502 showed a dose-dependent effect on IPS (supplemental Figure 5). Histopathologic analysis revealed that PB502 was superior to L-Kyn in reducing lung inflammation (Figure 7A). Although PB502 and L-Kyn were equally effective at reducing inflammatory cytokine levels in the lung (supplemental Figure 6A), PB502 was particularly effective in upregulating the immunomodulatory genes Il10, Tgfb1, Foxp3, and Entpd1 (Figure 7B). PB502 was also more effective at inhibiting IL-17A production and proliferation of CD4+ T cells in the lung during IPS (supplemental Figure 6B-D). These results indicate that PB502 suppresses Th17 cells but promotes the generation of regulatory T (Treg) cells. Indeed, administration of PB502 significantly increased the percentage of FoxP3+CD4+ Treg cells in the lung while decreasing CD44hi effector CD4+ T cells (Figure 7C). Delayed administration reproduced this pattern (Figure 7D), and in this experimental setting, CD4+ T cells isolated from the lung of PB502-injected recipients produced higher amounts of IL-10 compared with vehicle- or L-Kyn–injected recipients (Figure 7E). Next, we determined whether PB502 could directly promote Treg cell differentiation under 2 in vitro CD4+ T-cell culture conditions. First, PB502 exhibited synergy with TGF-β in driving the differentiation of CD4+ T cells toward FoxP3+ Treg cells (Figure 7F). This effect was more prominent in human CD4+ T-cell culture (Figure 7F). Second, although there was a mild effect of PB502 on mouse Treg differentiation under Th17 differentiation conditions, PB502 significantly increased the release of IL-10 (Figure 7G-H). PB502 induced a marked differentiation of human Treg cells under these conditions (Figure 7G). Increased PB502 concentration did not affect Treg cell differentiation when AHR was blocked (supplemental Figure 7). Finally, PB502 was effective at increasing overall survival and decreasing clinical symptoms (Figure 7I). The amelioration of GVHD was associated with a reduction in intestinal GVHD (supplemental Figure 8).
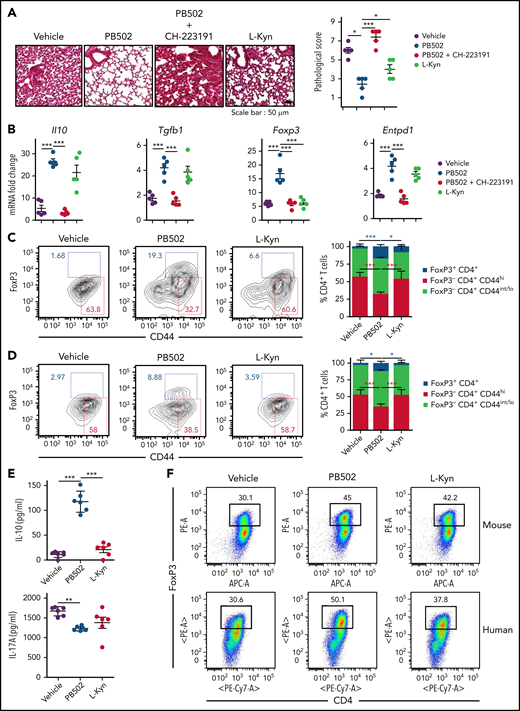

Therapeutic effects of PB502 on IPS. (A-D) Lethally irradiated B6.WT mice were injected with BALB/c.Ifng−/− T cells and BALB/c.WT TCD BM cells. (A-C) Recipients were intraperitoneally injected with PB502 (20 mg/kg) or L-Kyn (100 mg/kg) with or without CH-223191(10 mg/kg) each day from day 3 through day 8. Lungs were harvested on days 14 (A-B) and 21 (C). (A) Representative image of hematoxylin and eosin staining (×200; left) and pathologic scoring (right; n = 5 per group). (B) Levels of cytokine messenger RNA (mRNA; n = 5 per group). (C) Lung cells were stained for CD4, CD44, and Foxp3 and assessed by flow cytometry. Representative dot plots for Foxp3+ or CD44hi cells on gated CD4+ T cells (left) and composition of Foxp3+CD4+, Foxp3−CD44hiCD4+, and CD44int/loCD4+ T cells (right; n = 3-5 per group). (D-E) Recipient mice were intraperitoneally injected with PB502 (20 mg/kg) or L-Kyn (100 mg/kg) each day from day 7 through day 12. Lungs were harvested on day 27. (D) Data were analyzed and presented as described in panel C. (E) Isolated CD4+ T cells were restimulated with anti-CD3/CD28 for 24 hours, and the concentration of IL-10 and IL-17A was measured in the cell culture supernatants (n = 6 per group). (F) Naïve CD4+ T cells isolated from B6 splenocytes or human peripheral blood mononuclear cells were cultured under Treg differentiation conditions (anti-CD3/CD28, IL-2, and TGF-β) with PB502 (1 μM) or L-Kyn (100 μM) for 3 (mouse cells) or 5 days (human cells). Representative dot plots for Foxp3+CD4+ T cells are presented. (G-H) Naïve CD4+ T cells were cultured under Th17 differentiation conditions (anti-CD3/CD28, IL-6, TGF-β, IL-23, anti–IFN-γ, and anti–IL-4 for mouse cells and anti-CD3/CD28, TGF-β, IL-1β, and IL-23 for human cells) with PB502 (1 μM) or L-Kyn (100 μM) for 3 (mouse cells) or 5 days (human cells). (G) Representative dot plots for Foxp3+CD4+ T cells. (H) Concentrations of IL-10 in cell culture supernatants of mouse cells. (I) Lethally irradiated B6 mice were injected with BALB/c.Ifng−/− T cells (2 × 106) and BALB/c.WT TCD BM cells (1 × 107). Recipients were intraperitoneally injected with PB502 (20 mg/kg) or L-Kyn (100 mg/kg) each day from day 3 through day 16. Survival rates are presented at the left and clinical GVHD scores at the right. Results are representative of at least 3 independent experiments with similar results. Data represent mean ± standard error of the mean. One- or 2-way analysis of variance (A-H) and Mantel-Cox log-rank test (I) were performed. *P < .05, **P < .01, ***P < .001.
Therapeutic effects of PB502 on IPS. (A-D) Lethally irradiated B6.WT mice were injected with BALB/c.Ifng−/− T cells and BALB/c.WT TCD BM cells. (A-C) Recipients were intraperitoneally injected with PB502 (20 mg/kg) or L-Kyn (100 mg/kg) with or without CH-223191(10 mg/kg) each day from day 3 through day 8. Lungs were harvested on days 14 (A-B) and 21 (C). (A) Representative image of hematoxylin and eosin staining (×200; left) and pathologic scoring (right; n = 5 per group). (B) Levels of cytokine messenger RNA (mRNA; n = 5 per group). (C) Lung cells were stained for CD4, CD44, and Foxp3 and assessed by flow cytometry. Representative dot plots for Foxp3+ or CD44hi cells on gated CD4+ T cells (left) and composition of Foxp3+CD4+, Foxp3−CD44hiCD4+, and CD44int/loCD4+ T cells (right; n = 3-5 per group). (D-E) Recipient mice were intraperitoneally injected with PB502 (20 mg/kg) or L-Kyn (100 mg/kg) each day from day 7 through day 12. Lungs were harvested on day 27. (D) Data were analyzed and presented as described in panel C. (E) Isolated CD4+ T cells were restimulated with anti-CD3/CD28 for 24 hours, and the concentration of IL-10 and IL-17A was measured in the cell culture supernatants (n = 6 per group). (F) Naïve CD4+ T cells isolated from B6 splenocytes or human peripheral blood mononuclear cells were cultured under Treg differentiation conditions (anti-CD3/CD28, IL-2, and TGF-β) with PB502 (1 μM) or L-Kyn (100 μM) for 3 (mouse cells) or 5 days (human cells). Representative dot plots for Foxp3+CD4+ T cells are presented. (G-H) Naïve CD4+ T cells were cultured under Th17 differentiation conditions (anti-CD3/CD28, IL-6, TGF-β, IL-23, anti–IFN-γ, and anti–IL-4 for mouse cells and anti-CD3/CD28, TGF-β, IL-1β, and IL-23 for human cells) with PB502 (1 μM) or L-Kyn (100 μM) for 3 (mouse cells) or 5 days (human cells). (G) Representative dot plots for Foxp3+CD4+ T cells. (H) Concentrations of IL-10 in cell culture supernatants of mouse cells. (I) Lethally irradiated B6 mice were injected with BALB/c.Ifng−/− T cells (2 × 106) and BALB/c.WT TCD BM cells (1 × 107). Recipients were intraperitoneally injected with PB502 (20 mg/kg) or L-Kyn (100 mg/kg) each day from day 3 through day 16. Survival rates are presented at the left and clinical GVHD scores at the right. Results are representative of at least 3 independent experiments with similar results. Data represent mean ± standard error of the mean. One- or 2-way analysis of variance (A-H) and Mantel-Cox log-rank test (I) were performed. *P < .05, **P < .01, ***P < .001.
In our model where donor Ifng−/− T cells were used for IPS induction, donor Th17 CD4+ T cells mediated IPS pathogenesis, but donor CD8+ T cells were not required for induction of IPS, and PB502 had no distinguishable effect on lung inflammation (supplemental Figure 9). Because of this limitation of our model, we used the B6 → BDF1 acute GVHD model in which donor CD4+ and CD8+ T cells plays a pathogenic role in IPS34 to explore the therapeutic efficacy of PB502 in IPS. We found that PB502 decreased clinical symptoms of GVHD (supplemental Figure 10A) and significantly lowered histopathologic scores for IPS and gut GVHD but not for liver GVHD (supplemental Figure 10B). Levels of inflammatory cytokine genes reflected the extent of disease to the corresponding organs (supplemental Figure 10C). These results indicate that PB502 exerts a potent action on mucosal barrier organs but minimally affects systemic immunity. Indeed, PB502 and L-Kyn had no effect on proliferation or activation of donor T cells in the spleen or mesenteric lymph nodes (supplemental Figure 11A). In support of this, intranasal infusion of PB502 was effective at inhibiting IPS (supplemental Figure 11C-D).
Discussion
Barrier epithelial cell AHR has evolved to protect the body from environmental toxic substances, invading microbes, and injuries. Therefore, AHR adopts cell-intrinsic mechanisms linked to protective immunity and immune suppression, which prevents immunopathology. In the present study, we delineated the protective functions of AHR in lung epithelial cells during IPS. First, we showed that optimal expression of AHR in the lung requires preconditioning and donor T cells. Second, although lung epithelial cells and donor CD4+ T cells express high levels of AHR, the former are responsible for AHR-mediated regulation of the inflammatory response. Finally, an AHR-STAT1-JunD complex represses the AP-1–mediated transcriptional activation of Il6 and thereby functions as a roadblock in the differentiation of pathogenic Th17 cells. Our data suggest that endogenous signals transmitted through AHR in lung epithelial cells dampen the Th17-mediated inflammatory response during IPS. A lack of AHR causes hyperinflammation, whereas deliberate AHR activation with endogenous or synthetic ligands ameliorates the inflammatory profile in a mouse IPS model.
Two signals are required for AHR activation in lung epithelial cells: IFN-γ and IL-1β/other inflammatory cytokines to induce Ido1 and Ahr expression, respectively.12 Peaked expression of Ahr corresponds to the T-cell priming phase in the lung (Figure 1A), at which time AHR suppresses the expression of inflammatory cytokines, such as Il6, Tnf, and Il1b, in lung epithelial cells (Figure 3B). This suggests that AHR plays a central role in the cell-autonomous negative feedback regulation by inflammatory mediators during IPS. Donor T cells are required for AHR protein expression in the lung (Figure 1C). Although donor T cell–derived IFN-γ is not involved in the expression of Ahr, donor T cells seem to participate in maintaining stable AHR production by secreting other inflammatory cytokines, such as TNF-α (Figure 1G), or an as-yet-to be identified factor that helps the host immune cells produce inflammatory cytokines, such as IL-1β and IL-6.
In psoriasiform skin inflammation, upregulation of the AP-1 family of transcription factors occurs in Ahr−/− keratinocytes.20 Similarly, we found increased expression of AP-1 family genes, notably Jund, in lung epithelial cells after IPS induction (Figure 4C). We also determined how AHR represses transcriptional activation of Jund and, consequently, Il6. The results indicated that IL-1β–induced expression of Jund requires activation of STAT1 and, to a lesser extent, JunD and their binding to the Jund promoter (Figure 4G-H). However, activation of AHR by L-Kyn prevented STAT1 and JunD from binding to the Jund promoter (Figure 4I) by creating a complex consisting of AHR, STAT1, and JunD (Figure 6H). These results suggest that AHR blocks the transcriptional activation of STAT1 and JunD. Currently, it is unclear whether activated AHR inhibits STAT1 activation in the cytoplasm or whether it blocks the binding of STAT1 to the Jund promoter in the nucleus.35 It also remains to be determined whether an AHR/STAT1 complex represses inflammatory genes such as Il6.36
We identified a novel synthetic indole-3-acetamide analog (PB502) that is a potent AHR agonist (Figure 6) and displays therapeutic activity against IPS (Figure 7) and gut GVHD (supplemental Figure 8). Compared with L-Kyn, PB502 upregulates the immunomodulatory genes Il10, Tgfb1, Foxp3, and Entpd1 in the lung during IPS (Figure 7B) and increases FoxP3+ Treg cells (Figure 7C-H). Differentiation of Treg cells from either naïve CD4+ T cells or under conditions of Th17 polarization depends on AHR, as shown by others.37 AHR regulates FoxP3 expression directly37 and indirectly by regulating SMAD1.38 It may also be possible that PB502 promotes transdifferentiation of Th17 cells toward IL-10–producing type 1 Treg cells (Figure 7H).39 Thus, the superior therapeutic effect of PB502 on IPS may be linked to the generation of Treg cells and presumably type 1 Treg cells.
Patients with autosomal recessive IFN-γ receptor 2 or autosomal dominant IFN-γ receptor 1 deficiency have undergone successful allogeneic HSCT.40,41 In addition, IL-6, but not IFN-γ, has been shown to be produced by lung parenchyma and mediate IPS through induction of Th17 cells.11 Therefore, IFN-γ is unlikely to suppress IPS in patients who are administered a calcineurin inhibitor as GVHD prophylaxis. It remains enigmatic why IFN-γ cannot suppress IPS or vice versa in humans. This discrepancy is most likely a result of fundamental differences in mouse and human immunologies.42 There is evidence that control of STAT3 acetylation determines Ido1 transcription triggered by IL-6, which was linked directly to the regulation of IPS in mice that received Ifng−/− donor T cells.12 It has been suggested that the time window of action of calcineurin inhibitors is critical in early exacerbation of IPS; short-period posttransplantation administration of FK506 causes IPS to deteriorate but markedly increases long-term survival.12 Therefore, a testable explanation for the discrepancy between mouse and human is that the STAT3-IDO1 axis is more actively operated in patients under conditions in which IFN-γ expression is suppressed by administration with calcineurin inhibitors. Nonetheless, because there are increased levels of inflammatory cytokines in blood and bronchoalveolar lavage in patients with IPS,43-46 these cytokines may induce AHR expression during IPS, as suggested by our results. It is tempting to suggest that AHR is targetable for IPS therapy once AHR is confirmed to be expressed in the lung of patients with IPS. Indeed, indoles derived from gut microbiota display beneficial effects on murine gut GVHD by acting as AHR agonists, and decreased production of AHR ligands by microbiota is implicated in acute GVHD in humans.47-49 It will be interesting to test AHR agonists of diverse origins for their therapeutic efficacy in IPS and gut GVHD. However, caution is needed in applying AHR agonists to GVHD therapy, because AHR inhibits the maintenance of peripheral Treg cells during gut GVHD.50
In summary, our results suggest that impaired AHR signaling in lung epithelial cells after allogeneic HSCT leads to IPS development. Because lung epithelial cell AHR is upregulated during inflammatory conditions in an IFN-γ/IDO1–independent fashion, infusion of potent AHR agonists represents an effective strategy to block IPS without the requirement of IDO1 induction. The ability to induce the generation of Treg cells is an added benefit of our synthetic AHR agonist for treating IPS and gut GVHD. Our study provides an example of proof of concept–based drug development for IPS.
Acknowledgments
The authors thank Namdoo Kim (Voronoi) for giving advice on drug design.
This work was supported by National Research Foundation of Korea grants, funded by the Korean government, NRF-2016R1D1A1B03934016 and 2021R1A2C1010970 (S.-K.S.), NRF-2020M3A9D303789112 (B.K.), and 2018R1A6A3A01013228 (S.-M.L.).
Authorship
Contribution: S.-M.L., B.K., and S.-K.S. designed experiments; S.-M.L., C.E.K., H.Y.P., E.H.Y., H.J.W., J.M.A., N.Z.N.N., M.K., M.J., and S.P. performed experiments; W.H.J., W.-S.L., M.S.K., H.Y., S.W., and D.H.K. contributed to study design and data analysis; and S.-M.L., B.K., and S.-K.S. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Su-Kil Seo, Department of Microbiology and Immunology, College of Medicine, Inje University, Bokji-ro 75, Busanjin-gu, Busan 47392, Republic of Korea; e-mail; sseo@inje.ac.kr; and Byungsuk Kwon, School of Biological Sciences, University of Ulsan, Daehak-ro, Nam-gu, Ulsan 44610, Republic of Korea; e-mail: bkwon@ulsan.ac.kr.
For original data, please e-mail Su-Kil Seo at sseo@inje.ac.kr.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal