

Key Points
Antibody-mediated inhibition of MICA/B shedding promotes the phagocytosis of leukemia cells by macrophages.
Romidepsin synergizes with antibody-mediated inhibition of MICA/B shedding by inducing leukemia cells to upregulate the MICA/B expression.

Abstract
Acute myeloid leukemia (AML) is a clonal hematopoietic stem and progenitor cell malignancy characterized by poor clinical outcomes. Major histocompatibility complex class I polypeptide-related sequence A and B (MICA/B) are stress proteins expressed by cancer cells, and antibody-mediated inhibition of MICA/B shedding represents a novel approach to stimulate immunity against cancers. We found that the MICA/B antibody 7C6 potently inhibits the outgrowth of AML in 2 models in immunocompetent mice. Macrophages were essential for therapeutic efficacy, and 7C6 triggered antibody-dependent phagocytosis of AML cells. Furthermore, we found that romidepsin, a selective histone deacetylase inhibitor, increased MICB messenger RNA in AML cells and enabled subsequent stabilization of the translated protein by 7C6. This drug combination substantially increased surface MICA/B expression in a human AML line, pluripotent stem cell-derived AML blasts and leukemia stem cells, as well as primary cells from 3 untreated patients with AML. Human macrophages phagocytosed AML cells following treatment with 7C6 and romidepsin, and the combination therapy lowered leukemia burden in a humanized model of AML. Therefore, inhibition of MICA/B shedding promotes macrophage-driven immunity against AML via Fc receptor signaling and synergizes with an epigenetic regulator. These results provide the rationale for the clinical testing of this innovative immunotherapeutic approach for the treatment of AML.
Introduction
Acute myeloid leukemia (AML) has the highest fatality rate compared with other leukemias, with 20 240 and 11 400 new AML cases and deaths estimated to occur in the United States in 2021.1 Checkpoint blockade immunotherapy has revolutionized the care for other cancers, but anti-CTLA-4 and anti–PD-1 therapies have not demonstrated clinical benefit in AML trials.2-4 Somatic mutations can result in neoantigens essential for immunity triggered by anti-CTLA-4 and anti–PD-1; however, AML has overall low numbers of somatic mutations compared with other cancers that usually respond to checkpoint blockers.5,6 Chimeric antigen receptor (CAR) T cells recognize surface proteins, but the most common antigen in AML (ie, CD33) is also expressed by healthy myeloid cells (eg, Kupffer cells), and CD33-specific CAR T cells have been associated with severe hepatotoxicity.7 Therefore, immunotherapy for AML should (1) have mechanism independent of neoantigens and (2) be selective to malignant cells.
Malignant transformation triggers cellular stress pathways, which in turn increase expression of proteins that serve as danger signals. Two of them are major histocompatibility complex (MHC) class I polypeptide-related sequence A (MICA) and MHC class I polypeptide-related sequence B (MICB), here abbreviated as MICA/B.8 MICA/B are surface proteins on malignant cells and bind the natural killer group 2D (NKG2D) receptor, which stimulates natural killer (NK) cells and T cells.9 However, MICA/B are downregulated via proteolytic cleavage, a posttranslational modification that removes MICA/B from the cellular surface.10-14 The sera from patients with AML have average soluble MICA and MICB concentrations 10-fold higher than those from healthy donors.15 Accordingly, CAR T cells engineered to recognize NKG2D ligands were safe but did not generate antileukemia response in phase 1 trial; all patients with AML in this trial had soluble MICA, MICB, or both in blood plasmas.16 For these reasons, MICA/B shedding is a therapeutic target in AML.17
In a previous study, we developed the 7C6 monoclonal antibody (mAb) that binds MICA/B and inhibits cleavage without obstructing interaction with NKG2D. The 7C6 mAb restores the NKG2D recognition of tumor cells by increasing the cell surface density of MICA/B. It also triggers antibody-dependent cellular cytotoxicity via Fc receptors in NK cells. In melanoma models, 7C6 inhibits pulmonary metastases in a NK cell-dependent manner.18 For these reasons, we hypothesized that 7C6 promotes immunity also against AML. Here, we show that 7C6 inhibits the AML outgrowth by mechanism that is independent of NK cells. The 7C6 mAb increased surface MICA/B levels in leukemia cells, which in turn were phagocytosed by macrophages on Fc receptor engagement. We also show that romidepsin, an epigenetic therapeutic, further enhances the surface expression of MICA/B in leukemia cells when combined with 7C6. This drug combination inhibited leukemia in a humanized AML model. Therefore, immunotherapy for AML can be achieved with an antibody that increases the MICA/B stress markers in leukemia cells, which are then subsequently captured and destroyed by macrophages.
Methods
Tissue cultures
NB4 was donated by Zhen-Qiang Pan (Mount Sinai), whereas C1498 and WEHI-3 were purchased from American Type Culture Collection. All 3 tested negative for mycoplasma and rodent pathogens. Induced pluripotent stem cell (iPSC)-derived AML blasts and leukemia stem cells (LSCs) were generated through in vitro differentiation of the AML-iPSC line AML-4.10, as reported previously.19,20 Cryopreserved peripheral blood mononuclear cells from deidentified patients with AML were provided by the Hematological Malignancies Tissue Bank (Mount Sinai), which has institutional review board approval (protocol #11-1669(0001)(01)).
The lentiviral vector used to transduce C1498 and WEHI-3 is in a public repository (Addgene, catalog no. 114008) and was reported.18 C1498-MICB and WEHI-3-MICB were cultured in Dulbecco’s modified Eagle medium complemented with 10% fetal bovine serum, Glutamax, Penicillin/Streptomycin, and N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid. NB4 was cultured in RPMI-1640 complemented as previously. Primary cells were cultured in StemSpan (StemCell Technologies) complemented with 150 ng/mL stem cell factor, 100 ng/mL thrombopoietin, 100 ng/mL Fms-related tyrosine kinase 3 ligand, and 50 ng/mL interleukin-3. Cells were cultured at 37°C and 5% CO2.
Gene expression analyses
NB4, C1498-MICB, and WEHI-3-MICB had messenger RNAs (mRNAs) isolated with RNeasy Plus Mini Kit (QIAGEN), which includes genomic DNA Eliminator Columns. Complementary DNA (cDNA) was synthesized with ProtoScript Kit (New England BioLabs), and polymerase chain reaction (PCR) performed with DreamTaq Green Master Mix (Thermo Fisher Scientific). The primer pair sequences are in supplemental Table 1, available on the Blood Web site. Following PCR, analyses were done via electrophoresis and ImageJ, with normalization by the housekeeping gene. Murine AML lines were also analyzed by flow cytometry with Fc receptor blockade (mouse TruStain FcX) and labeling with Zombie Yellow (Biolegend) plus anti-MULT-1 (237104), anti-pan Rae-1 (18610), or anti-H60 (205326) (R&D Systems).
For MICA/B gene expression analyses, NB4 was treated for 24 hours with 10 nmol/L of the inhibitors in supplemental Table 2, all dissolved in dimethyl dioxide, which was used as control. Gene expression was analyzed as previously.
MICA/B shedding assays
Cells were cultured for 24 hours in 96-well U-bottom plates. NB4, C1498-MICB, and WEHI-3-MICB had supernatants collected and soluble MICA and MICB were analyzed by Human MICA or MICB DuoSet ELISA kits (R&D Systems). These cells were analyzed by flow cytometry with Fc receptor blockade (mouse/human TruStain FcX), and labeling with anti-MICA/B (6D4) and Zombie Yellow, from BioLegend. Staining with murine NKG2D-Fc chimera (R&D Systems) was revealed by human immunoglobulin G (IgG) Fc antibody (M1310G05). Primary cells from patients with AML were also labeled with anti-CD45 (2D1), anti-CD33 (P67.6), and anti-CD34 (561). All analyses, including those that follow, were done in a BD LSRII Fortessa followed by FlowJo v10.
Phagocytosis assays
C1498-MICB and NB4 were pretreated with 20 μg/mL of antibodies (7C6-mIgG2a, 7C6-DANA, 7C6-hIgG1, or isotypes) for 24 hours; NB4 was also pretreated with 0 or 5 nmol/L romidepsin. Subsequently, these cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE; BioLegend) and cocultured for 1 hour with bone marrow-derived (mouse) or monocyte-derived (human) macrophages differentiated via macrophage colony-stimulating factor. Subsequently, mouse and human cell suspensions were labeled with anti-F4/80 (BM8) or anti-CD14 (63D3), respectively.
Mouse AML models
All procedures, including the human AML model, were approved by the institutional animal care and use committee. Mice were 6 to 8 weeks old and from The Jackson Laboratory. Male C57BL/6 mice were used in the C1498-MICB model, except in an experiment with female C57BL/6 mice. Male Balb/c mice were used in the WEHI-3-MICB model. A total of 2 × 106 leukemia cells were inoculated IV via tail vein. On days 5 and 6, unless otherwise indicated, mice received intraperitoneal (IP) injections of 200 μg of 7C6-mIgG2a, 7C6-mIgG2b-DANA, or isotype (C1184, BioXcell). On day 14, mice received 100 μg of antibodies. Euthanasia via CO2 and analyses of leukemia in the bloods and femoral bone marrows were performed approximately 3 weeks after leukemia inoculation (the precise days are indicated in figure legends). In the survival experiments, 1 × 106 C1498-MICB or 5 × 105 WEHI-3-MICB were injected and mice were euthanized when they developed physical signs of disease (apparent loss of body weight, moribund state, limb paralysis, or curved posture).
Analysis of leukemia burden in the blood and femoral bone marrow was performed by flow cytometry upon staining with Fc receptor blockers, anti-CD45.2 (104), and Zombie Yellow. For analysis of bone marrow and spleen macrophages, anti-F4/80 (BM8), anti-CD16/CD32 (93), and anti-CD64 (X54-5/7.1) were used. To determine the absolute numbers of leukemia cells, blood samples, and femoral bone marrows were lysed with ACK buffer (Gibco) and cells were counted in hemocytometer. The percentages of ZsGreen+ leukemia cells were used to calculate the number of leukemia cells per milliliter of blood and total number of leukemia cells per femoral bone marrow.
In mechanistic experiments, anti-NK1.1 (PK136), anti-CD8β (5358), anti-γδTCR (UC7-13D5), anti-CD1d (20H2-HB323), anti-NKG2D (HMG2D), or control IgG (MOPC-21), all from BioXcell, were inoculated intraperitoneally (100 μg per mouse) on days −1, 0, 7, and 14. For macrophage depletion, 0.2 mL clodronate or control liposomes (Encapsula NanoSciences) were inoculated IV on days −1, 5, and 14.
Human AML model
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were 6 to 8 weeks old, female, and from The Jackson Laboratory. NSG mice were inoculated IV with 2 × 106 NB4 cells. On day 23, a subset of mice underwent submandibular bleeding. On days 23 and 24, romidepsin at doses of 10, 5, 2.5, or 0 mg/kg was administered via IP injections, together with 0.2 mg of 7C6-hIgG1-DANA. Analyses of surface MICA/B were performed on day 25 by flow cytometry upon staining with Fc Receptor blockers, anti-mouse CD45.1 (A20), anti-human CD45 (2D1), anti-human CD33 (P67.6), anti-human MICA/B (6D4), and Zombie Yellow. For analyses of leukemia burdens, treatments with 0.2 mg of 7C6-mIgG2a or isotype (C1184) and romidepsin at 0, 2.5, or 5 mg/kg were given on days 18 and 19; analyses of leukemia were performed as previously on day 25.
Statistical analyses
Data are the mean ± standard error (SE) or deviation (SD) or median ± interquartile range. The statistical tests were Mann-Whitney U, 2-way analysis of variance with Bonferroni’s test, and logrank, as indicated in figure legends. P < .05 was considered statistically significant. Statistics were performed with GraphPad Prism 9.
Results
7C6 inhibits AML in immunocompetent mice
Mice and humans have NKG2D, but the ligands have distant homology between these species.8 To mimic the immunoescape by MICA/B shedding, we transduced murine AML cell lines with lentiviral vector that encodes human MICB. This construct has the cDNA sequence of MICB*005, the most frequent polymorphic variant in humans.21
C1498 is a cell line of murine AML that arose spontaneously into a C57BL/6 mouse.22 We infected C1498 cells with the lentivirus that drives MICB expression. C1498-MICB expresses the myeloperoxidase (Mpo) gene; although the expression level is low, it is consistent with AML designation (supplemental Figure 1A). C1498-MICB also expresses an endogenous NKG2D ligand (supplemental Figure 1B). The 7C6 mAb (with murine IgG2b heavy chain, mIgG2b) inhibits the MICB release into supernatants of C1498-MICB cultures and stabilizes the surface protein (Figure 1A; supplemental Figure 1C-E). We IV inoculated C57BL/6 mice with C1498-MICB and, 5 days later, treated them with 7C6 (murine IgG2a, mIgG2a) or isotype. Euthanasia and analyses of leukemia cells were performed 3 weeks following C1498-MICB inoculation. The lentiviral vector also has ZsGreen for ex vivo identification of C1498-MICB in the blood and bone marrow (Figure 1B; supplemental Figure 2A-B). The 7C6-mIgG2a lowers the percentages of C1498-MICB to almost undetectable levels in the blood and femoral bone marrow (Figure 1C). In absolute numbers, 7C6-mIgG2a decreases by ∼10-fold the concentrations and numbers of C1498-MICB cells in the blood and femoral bone marrow, respectively, compared with isotype (Figure 1D). Histopathology revealed large nucleated cells in the blood and expansion of large cells with basophilic cytoplasm in the femoral bone marrow, respectively, from isotype-treated mice but not in those from 7C6-mIgG2a-treated or naïve mice (Figure 1E). The 7C6-mIgG2a prolonged the survival of leukemic mice, as analyzed in separate experiments with mice being euthanized when they develop physical signs of disease (Figure 1F). Notably, all these experiments had male mice (Figure 1B-F), but C1498 arose in a female mouse.22 To mitigate this discrepancy, we confirmed that 7C6-mIgG2a inhibits C1498-MICB also in female mice but this cell line develops less leukemia (supplemental Figure 3A-B), so we restricted the C1498-MICB model to male mice only.

7C6 inhibits the in vivo outgrowth of AML in 2 mouse models. (A-F) C1498-MICB and AML model in immunocompetent C57BL/6 mice. (G-J) WEHI-3-MICB and AML model in immunocompetent Balb/c mice. (A) 7C6-mIgG2b inhibits the shedding of MICB by C1498 cells. The C1498-MICB cell line was cultured for 24 hours with the indicated antibodies at the indicated doses, followed by analysis of soluble MICB shed into supernatants with a sandwich enzyme-linked immunosorbent assay (ELISA) and surface MICB on cells via flow cytometry. MFI, mean fluorescence intensity. (B-E) C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB cells, which corresponds to day 0. Mice were treated via IP injections with 200 μg of the indicated antibodies on days 5 and 6; on day 14, mice were treated with just 100 μg of the indicated antibodies. Euthanasia and analysis of blood and bone marrow were performed on day 21. (B) Illustration of the procedure and identification of ZsGreen+ C1498-MICB in the blood by flow cytometry. (C-E) 7C6-mIgG2a reduces the frequency (C) and absolute numbers (D) of C1498-MICB in the blood and bone marrow of mice. (E) Similar results were found via histopathology. (C) The frequency of C1498-MICB was measured after staining blood cells with an antigen-presenting cell-conjugated CD45 antibody followed by identification of ZsGreen+ C1498-MICB by flow cytometry. (D) The absolute numbers of C1498-MICB were determined after multiplying the C1498-MICB frequency by the concentration of total blood cells and bone marrow cells from femur. Wright-Giemsa staining (top) and hematoxylin-eosin staining (bottom) of blood smears and femurs, respectively. (E) The arrows indicate leukemia-like cells. (F) 7C6-mIgG2a prolongs the survival of leukemia-bearing mice. C57BL/6 mice were inoculated IV with 1 × 106 C1498-MICB cells and treated with the indicated antibodies on days 5, 6, and 14 (same antibody treatment regimen of experiment shown in panels C-E). Mice were euthanized when they showed physical signs of disease (weight loss, limb paralysis, moribund state, and/or curved posture). (G) 7C6-mIgG2a inhibits the MICB shedding by WEHI-3-MICB and stabilizes the surface protein. WEHI-3-MICB was treated for 24 hours with the indicated concentrations of the indicated antibodies, followed by analyses of soluble MICB shed into supernatants by sandwich ELISA and surface MICB on cells by flow cytometry. (H-I) 7C6 inhibits WEHI-3-MICB in vivo. Balb/c mice were inoculated IV with 2 × 106 WEHI-3-MICB and, on days 5 and 6, treated with 0.2 mg of 7C6-mIgG2a or isotype. On day 14, only 0.1 mg of antibodies were administered. Euthanasia and analyses of leukemia by flow cytometry were done on day 20. (J) 7C6 prolongs the survival of mice in the WEHI-3-MICB model. Balb/c mice were inoculated IV with 5 × 105 WEHI-3-MICB and treated with 0.2 mg of 7C6-mIgG2a or isotype on days 1, 4, and 8. As in panel F, mice were euthanized when they showed physical signs of disease. Data representative of 3 (A,G) and pooled of 2 (C-D,F,H-J) independent experiments. Data in panel E are representative of 5 mice per antibody and 2 naïve mice. (C-D,H-I) **P < .01; ***P < .001, (Mann-Whitney test). In panels F and J, the P values were calculated by Mantel-Cox test. In panels A and G, 3 wells per antibody concentration were used. (C-D,H-I) Each dot indicates 1 mouse. Data are (A,G) mean ± standard error or (C-D,H-I) median ± interquartile range.
7C6 inhibits the in vivo outgrowth of AML in 2 mouse models. (A-F) C1498-MICB and AML model in immunocompetent C57BL/6 mice. (G-J) WEHI-3-MICB and AML model in immunocompetent Balb/c mice. (A) 7C6-mIgG2b inhibits the shedding of MICB by C1498 cells. The C1498-MICB cell line was cultured for 24 hours with the indicated antibodies at the indicated doses, followed by analysis of soluble MICB shed into supernatants with a sandwich enzyme-linked immunosorbent assay (ELISA) and surface MICB on cells via flow cytometry. MFI, mean fluorescence intensity. (B-E) C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB cells, which corresponds to day 0. Mice were treated via IP injections with 200 μg of the indicated antibodies on days 5 and 6; on day 14, mice were treated with just 100 μg of the indicated antibodies. Euthanasia and analysis of blood and bone marrow were performed on day 21. (B) Illustration of the procedure and identification of ZsGreen+ C1498-MICB in the blood by flow cytometry. (C-E) 7C6-mIgG2a reduces the frequency (C) and absolute numbers (D) of C1498-MICB in the blood and bone marrow of mice. (E) Similar results were found via histopathology. (C) The frequency of C1498-MICB was measured after staining blood cells with an antigen-presenting cell-conjugated CD45 antibody followed by identification of ZsGreen+ C1498-MICB by flow cytometry. (D) The absolute numbers of C1498-MICB were determined after multiplying the C1498-MICB frequency by the concentration of total blood cells and bone marrow cells from femur. Wright-Giemsa staining (top) and hematoxylin-eosin staining (bottom) of blood smears and femurs, respectively. (E) The arrows indicate leukemia-like cells. (F) 7C6-mIgG2a prolongs the survival of leukemia-bearing mice. C57BL/6 mice were inoculated IV with 1 × 106 C1498-MICB cells and treated with the indicated antibodies on days 5, 6, and 14 (same antibody treatment regimen of experiment shown in panels C-E). Mice were euthanized when they showed physical signs of disease (weight loss, limb paralysis, moribund state, and/or curved posture). (G) 7C6-mIgG2a inhibits the MICB shedding by WEHI-3-MICB and stabilizes the surface protein. WEHI-3-MICB was treated for 24 hours with the indicated concentrations of the indicated antibodies, followed by analyses of soluble MICB shed into supernatants by sandwich ELISA and surface MICB on cells by flow cytometry. (H-I) 7C6 inhibits WEHI-3-MICB in vivo. Balb/c mice were inoculated IV with 2 × 106 WEHI-3-MICB and, on days 5 and 6, treated with 0.2 mg of 7C6-mIgG2a or isotype. On day 14, only 0.1 mg of antibodies were administered. Euthanasia and analyses of leukemia by flow cytometry were done on day 20. (J) 7C6 prolongs the survival of mice in the WEHI-3-MICB model. Balb/c mice were inoculated IV with 5 × 105 WEHI-3-MICB and treated with 0.2 mg of 7C6-mIgG2a or isotype on days 1, 4, and 8. As in panel F, mice were euthanized when they showed physical signs of disease. Data representative of 3 (A,G) and pooled of 2 (C-D,F,H-J) independent experiments. Data in panel E are representative of 5 mice per antibody and 2 naïve mice. (C-D,H-I) **P < .01; ***P < .001, (Mann-Whitney test). In panels F and J, the P values were calculated by Mantel-Cox test. In panels A and G, 3 wells per antibody concentration were used. (C-D,H-I) Each dot indicates 1 mouse. Data are (A,G) mean ± standard error or (C-D,H-I) median ± interquartile range.
The WEHI-3 AML line arose in a male Balb/c mouse treated with carcinogenic cocktail.23 We transduced this cell line with our lentivirus. WEHI-3-MICB expresses Mpo and it expresses H60 and low levels of Rae-1 that are endogenous NKG2D ligands (supplemental Figure 4A-B). WEHI-3-MICB sheds high levels of MICB into culture supernatants, yet 7C6-mIgG2a inhibits the shedding and stabilizes the surface protein (Figure 1G; supplemental Figure 4C-E). We used WEHI-3-MICB as an AML model in immunocompetent male Balb/c mice. Treatments with 7C6-mIgG2a or isotype started on day 5 and euthanasia, followed by flow cytometry, were performed 3 weeks after WEHI-3-MICB inoculation. The 7C6-mIgG2a lowered the percentages of WEHI-3-MICB to almost undetectable levels in the blood and femoral bone marrow (Figure 1H). The absolute numbers of leukemia cells in both tissues were reduced by 100-fold (Figure 1I). The 7C6-mIgG2a also prolonged mouse survival in the WEHI-3-MICB model (Figure 1J). Altogether, these data indicate that the inhibition of MICB shedding with 7C6 potently inhibits AML outgrowth.
Importance of NK cells, CD8 T cells, γδT cells, NKT cells, and NKG2D for the ability of 7C6 in inhibiting AML
MICA/B have no intracellular signaling domains nor associate with adaptor molecules that transmit signals, and thereby the inhibition of shedding by itself does not kill tumor cells.8,18 In previous studies with melanoma models, we found that 7C6 inhibits metastases in a NK cell-dependent manner.18,24 For this reason, we hypothesized that NK cells are also involved in the AML models. Anti-NK1.1 (α-NK1.1) depletes NK cells in vivo.18,24,25 Mice treated with α-NK1.1 have an increase in leukemia burden, but surprisingly still respond to 7C6-mIgG2a. The percentages of C1498-MICB in the blood and bone marrow were extremely low in mice cotreated with 7C6-mIgG2a and α-NK1.1 (Figure 2A). NK cell depletion increased the concentrations and total numbers of AML cells in the blood and bone marrow, but 7C6-mIgG2a still lowered the numbers of AML cells by ∼10-fold compared with isotype (Figure 2B). These results lead us to question if NK cells were depleted. We validated the NK cell depletion by identifying blood NK cells before, but not after, α-NK1.1 in a separate cohort of healthy mice (Figure 2C). Therefore, the inhibition of AML by 7C6-mIgG2a is optimal with NK cells but is not primarily driven by them.
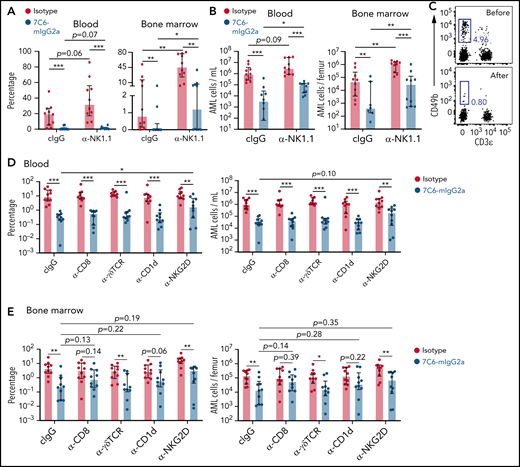
The importance of several lymphocyte populations for the ability of 7C6 in inhibiting AML. (A-B) Antibody-mediated depletion of NK cells does not abrogate the 7C6’s efficacy against AML. C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB (day 0) and treated via IP injections with 200 μg 7C6-mIgG2a or isotype on days 5 and 6. On day 14, mice were treated with just 100 μg of 7C6-mIgG2a or isotype. Furthermore, mice were treated with 100 μg control IgG (cIgG) or α-NK1.1 on days −1, 0, 7, and 14. Analysis of leukemia cells in the blood and bone marrow were performed on day 20 by flow cytometry. (A) Frequency of C1498-MICB in the blood and bone marrow. (B) Absolute numbers of C1498-MICB in the blood and bone marrow. (C) Validation of NK cell depletion. A healthy mouse was bled via submandibular puncture and treated with 0.1 mg of α-NK1.1. Two days later, the mouse was euthanized and its blood collected for analyses. NK cells are the CD49b+ and CD3ε− cells. The gatings of lymphocytes, doublets discrimination, and identification of viable CD45.2+ cells (not shown) antecede the plots shown in panel C. (D-E) Antibody-mediated depletion/blockade of T cells and NKG2D. C57BL/6 mice were treated with the indicated depleting/blocking antibodies on days −1, 0, 7, and 14. The mice were also inoculated IV with 2 × 106 C1498-MICB (day 0) and treated with 0.2 mg 7C6-mIgG2a or isotype control on days 5 and 6. On day 14, mice received only 0.1 mg of 7C6-mIgG2a or isotype. Analyses of leukemia cells (D) in the blood and (E) bone marrow were done by flow cytometry 3 weeks after leukemia cell inoculation. Data pooled from (A-B,D-E) 2 independent experiments or (C) representative of 2 mice and 2 independent experiments. *P < .05; **P < .01; ***P < .001, as calculated (A-B,D-E) with Mann-Whitney tests comparing 2 treatment groups at a time. (A-B,D-E) Median ± interquartile range.
The importance of several lymphocyte populations for the ability of 7C6 in inhibiting AML. (A-B) Antibody-mediated depletion of NK cells does not abrogate the 7C6’s efficacy against AML. C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB (day 0) and treated via IP injections with 200 μg 7C6-mIgG2a or isotype on days 5 and 6. On day 14, mice were treated with just 100 μg of 7C6-mIgG2a or isotype. Furthermore, mice were treated with 100 μg control IgG (cIgG) or α-NK1.1 on days −1, 0, 7, and 14. Analysis of leukemia cells in the blood and bone marrow were performed on day 20 by flow cytometry. (A) Frequency of C1498-MICB in the blood and bone marrow. (B) Absolute numbers of C1498-MICB in the blood and bone marrow. (C) Validation of NK cell depletion. A healthy mouse was bled via submandibular puncture and treated with 0.1 mg of α-NK1.1. Two days later, the mouse was euthanized and its blood collected for analyses. NK cells are the CD49b+ and CD3ε− cells. The gatings of lymphocytes, doublets discrimination, and identification of viable CD45.2+ cells (not shown) antecede the plots shown in panel C. (D-E) Antibody-mediated depletion/blockade of T cells and NKG2D. C57BL/6 mice were treated with the indicated depleting/blocking antibodies on days −1, 0, 7, and 14. The mice were also inoculated IV with 2 × 106 C1498-MICB (day 0) and treated with 0.2 mg 7C6-mIgG2a or isotype control on days 5 and 6. On day 14, mice received only 0.1 mg of 7C6-mIgG2a or isotype. Analyses of leukemia cells (D) in the blood and (E) bone marrow were done by flow cytometry 3 weeks after leukemia cell inoculation. Data pooled from (A-B,D-E) 2 independent experiments or (C) representative of 2 mice and 2 independent experiments. *P < .05; **P < .01; ***P < .001, as calculated (A-B,D-E) with Mann-Whitney tests comparing 2 treatment groups at a time. (A-B,D-E) Median ± interquartile range.
NKG2D is also expressed by T cells and provides costimulation to CD8 T cells.9,26-28 To establish the importance of T cells for the in vivo activity of 7C6, we used a panel of antibodies that are known to deplete CD8 T cells and γδT cells or that inhibit NKT cell-driven immunity by blocking CD1d.29-31 We also used a NKG2D antibody that blocks interaction with MICB (supplemental Figure 5). None of these depleting/blocking antibodies stopped 7C6 from inhibiting AML in the blood, although anti-NKG2D increased the percentage of leukemia cells in 7C6-treated mice (Figure 2D). In contrast, the effects in the bone marrow were more variable, with anti-γδTCR and anti-NKG2D having no impact, whereas anti-CD8 and CD1d evened up the degrees of leukemia burdens in the isotype and 7C6-mIgG2a groups (Figure 2E). Therefore, the inhibition of AML by 7C6 is optimal in the presence of these T-cell populations but it does not appear to be primarily mediated by them.
Macrophages inhibit AML by phagocytosing the 7C6-tagged leukemia cells
Tissue resident macrophages capture and digest circulating leukemia cells via phagocytosis.32 The 7C6-mIgG2a has Fc domain known to bind with high affinity to activating receptors that trigger antibody-dependent phagocytosis.33 We identified CD11b+ F4/80+ macrophages in the spleen and bone marrow from mice, whereas inoculation of clodronate liposomes successfully depleted these cells (Figure 3A; supplemental Figure 6A-B). These macrophages acquire ZsGreen fluorescence in mice inoculated with C1498-MICB, indicating that they phagocytose fluorescent leukemia cells (Figure 3B; supplemental Figure 6C). Spleen and bone marrow macrophages express high levels of CD16/CD32 and CD64, which are Fc-activating receptors (Figure 3C).

7C6 inhibits AML by triggering antibody-dependent phagocytosis. (A) Identification of macrophages and validation of their depletion in the spleen and bone marrow. C57BL/6 mice were inoculated IV with 0.2 mL of control or clodronate liposomes on days −8 and −1. On day 0, spleens and femoral bone marrows were collected after euthanasia, and CD11b+ F4/80+ macrophages, which are also negative for CD49b, CD3ε, B220, and Ly6G (supplemental Figure 6A-B), were analyzed by flow cytometry. Data representative of 2 mice per experiment and 2 independent experiments. (B-C) Characterization of spleen and bone marrow macrophages from leukemic mice. C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB and euthanized on day 20 for analyses. Naïve mice did not receive the leukemia cells and were euthanized together with the leukemic mice. Macrophages were identified by flow cytometry as in panel A. Spleen and bone marrow macrophages (B) acquire the ZsGreen fluorescence and (C) express high levels of Fc activating receptors in mice that were inoculated with C1498-MICB. (C) Red = isotype, Blue = anti-CD16/32 or anti-CD64. (D) Macrophages are required by 7C6 to inhibit AML. C57BL/6 mice were inoculated IV with 0.2 mL of clodronate or control liposomes on days −1, 5, and 14, and with 2 × 106 C1498-MICB on day 0. Antibody treatments were performed on days 5, 6, and 14, and euthanasia for flow cytometry-based analyses of leukemia burdens on day 19. (E) 7C6-mIgG2a triggers antibody-dependent phagocytosis. C57BL/6 mice’s bone marrow cells were differentiated to macrophages via 7 to 14 days culture with 10 ng/mL macrophage colony-stimulating factor. Before assay, C1498-MICB cells were treated with 20 μg/mL of 7C6-mIgG2a, 7C6-DANA, or isotype. Subsequently, these leukemia cells were labeled with CFSE and cocultured at 37°C or just incubated at 4°C with macrophages for 1 hour. After this, macrophages were detached with 10 mmol/L EDTA, labeled with F4/80 antibody, and analyzed by flow cytometry. (F) Fc receptor recognition is required by 7C6 to inhibit AML. Frequency (left) and absolute numbers (right) of leukemia cells are shown. C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB and treated with antibodies on days 5, 6, and 14. Euthanasia and flow cytometry-based analyzes of leukemia cells in the blood and bone marrow were done on day 19. Data are (B,D,F) median ± interquartile range or (E) mean ± SD, and are representative of (A) 2 or (E) 3, or (B,D,F) pooled of 2 independent experiments. *P < .05; **P < .01; ***P < .001, Mann-Whitney test comparing (B,D,F) 2 groups or (E) 2-way ANOVA with Bonferroni’s post hoc test.
7C6 inhibits AML by triggering antibody-dependent phagocytosis. (A) Identification of macrophages and validation of their depletion in the spleen and bone marrow. C57BL/6 mice were inoculated IV with 0.2 mL of control or clodronate liposomes on days −8 and −1. On day 0, spleens and femoral bone marrows were collected after euthanasia, and CD11b+ F4/80+ macrophages, which are also negative for CD49b, CD3ε, B220, and Ly6G (supplemental Figure 6A-B), were analyzed by flow cytometry. Data representative of 2 mice per experiment and 2 independent experiments. (B-C) Characterization of spleen and bone marrow macrophages from leukemic mice. C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB and euthanized on day 20 for analyses. Naïve mice did not receive the leukemia cells and were euthanized together with the leukemic mice. Macrophages were identified by flow cytometry as in panel A. Spleen and bone marrow macrophages (B) acquire the ZsGreen fluorescence and (C) express high levels of Fc activating receptors in mice that were inoculated with C1498-MICB. (C) Red = isotype, Blue = anti-CD16/32 or anti-CD64. (D) Macrophages are required by 7C6 to inhibit AML. C57BL/6 mice were inoculated IV with 0.2 mL of clodronate or control liposomes on days −1, 5, and 14, and with 2 × 106 C1498-MICB on day 0. Antibody treatments were performed on days 5, 6, and 14, and euthanasia for flow cytometry-based analyses of leukemia burdens on day 19. (E) 7C6-mIgG2a triggers antibody-dependent phagocytosis. C57BL/6 mice’s bone marrow cells were differentiated to macrophages via 7 to 14 days culture with 10 ng/mL macrophage colony-stimulating factor. Before assay, C1498-MICB cells were treated with 20 μg/mL of 7C6-mIgG2a, 7C6-DANA, or isotype. Subsequently, these leukemia cells were labeled with CFSE and cocultured at 37°C or just incubated at 4°C with macrophages for 1 hour. After this, macrophages were detached with 10 mmol/L EDTA, labeled with F4/80 antibody, and analyzed by flow cytometry. (F) Fc receptor recognition is required by 7C6 to inhibit AML. Frequency (left) and absolute numbers (right) of leukemia cells are shown. C57BL/6 mice were inoculated IV with 2 × 106 C1498-MICB and treated with antibodies on days 5, 6, and 14. Euthanasia and flow cytometry-based analyzes of leukemia cells in the blood and bone marrow were done on day 19. Data are (B,D,F) median ± interquartile range or (E) mean ± SD, and are representative of (A) 2 or (E) 3, or (B,D,F) pooled of 2 independent experiments. *P < .05; **P < .01; ***P < .001, Mann-Whitney test comparing (B,D,F) 2 groups or (E) 2-way ANOVA with Bonferroni’s post hoc test.
The 7C6-mIgG2a does not inhibit AML in mice inoculated with clodronate liposomes (Figure 3D; supplemental Figure 7). In phagocytosis assays, <20% of macrophages capture (at 4°C), and then ingest (at 37°C), isotype-treated leukemia cells, whereas ∼50% and 30% of macrophages capture and ingest, respectively, 7C6-mIgG2a-treated leukemia cells (Figure 3E; supplemental Figure 8A-B).
We have developed a variant of 7C6 that has D265A N927A (DANA) mutations, which abolish binding to Fc receptors.18 The 7C6-DANA stabilizes surface MICB in C1498-MICB (supplemental Figure 9), but 7C6-DANA does not trigger phagocytosis (Figure 3E; supplemental Figure 8B) nor does it inhibit AML in vivo (Figure 3F). Therefore, 7C6-mIgG2a inhibits AML by triggering antibody-dependent phagocytosis.
Romidepsin and 7C6 synergize to increase MICA/B in human AML and promote phagocytosis
MICB is constitutively expressed by the murine AML lines, but human cancers express MICA/B in response to cellular stress.8 However, ∼50% of AML patients have leukemia cells that lack MICA/B.34,35 To bypass these limitations and broaden the applicability of 7C6, we screened epigenetic therapies that increase MICA/B mRNA expressions. We used the NB4 line, which was isolated from a female patient with acute promyelocytic leukemia who relapsed after chemotherapy. NB4 and fresh blasts from the original patient have the t(15;17) chromosomal translocation; NB4 tests positive for myeloperoxidase reaction and expresses CD33 but not CD34.36
We first attempted to increase MICA/B expression with hypomethylating agents, but after performing several preliminary experiments, we concluded that azacitidine and decitabine do not increase MICA/B expression in NB4 or primary cells (not shown). Previous studies have shown that histone deacetylase (HDAC) inhibitors induce malignant cells to express MICA/B.37-40 To develop an alternative approach, we rationally screened a panel of selective inhibitors with minimum overlap of specificity for the class 1 HDACs (supplemental Table 2). Panobinostat, a pan HDAC inhibitor, was used for comparison. These inhibitors were given at a relatively low dose of 10 nmol/L to NB4 because our goal was to identify selective HDAC inhibitors that increase MICA/B mRNA at low doses that may cause minimum toxicity if administered in vivo. We discovered that romidepsin, an HDAC 1- and 2-specific inhibitor, increases MICB mRNA in NB4 cells. All other inhibitors, including mocetinostat that also inhibits HDACs 1 and 2 and panobinostat that has broad specificity, did not increase MICA/B mRNA at the tested dose (Figure 4A).

An HDAC 1 and 2-specific inhibitor, romidepsin, synergizes with 7C6 to increase the surface MICA/B expression levels. (A) Romidepsin increases MICB mRNA expression. NB4 cells were treated for 24 hours with 10 nmol/L of the indicated inhibitors or equal volume of dimethyl dioxide. Subsequently, mRNA was extracted, cDNA was synthetized, and PCR was performed with primers specific for the indicated genes plus GAPDH. Analyses were performed via electrophoresis and ImageJ software with GAPDH normalization. (B-C) Romidepsin increases MICB expression for stabilization of the surface protein by 7C6. NB4 cells were treated for 24 hours with 20 μg/mL of the indicated antibodies plus the indicated concentrations of romidepsin. Subsequently, soluble MICA/B shed into culture supernatants were analyzed by sandwich ELISA kits specific for either MICA or MICB. Surface MICA/B and cellular viability were analyzed by flow cytometry upon labeling of cells with 6D4 and Zombie Yellow, respectively. (D) Romidepsin + 7C6 treatment of leukemia cells enhances phagocytosis. NB4 cells were treated for 24 hours with romidepsin or equal volume of phosphate-buffered saline + 20 μg/mL of the indicated antibodies, and subsequently labeled with CFSE. Monocyte-derived macrophages from healthy donors were cocultured for 1 hour with the treated NB4 cells, and phagocytosis was analyzed by flow cytometry via identification of CD14+ CFSE+ macrophages. (E) Romidepsin and 7C6 increase the surface MICA/B expression levels in iPSC-derived AML blasts and LSC. The indicated cells were treated for 24 hours with the indicated concentrations of romidepsin plus 20 μg/mL of the indicated antibodies, and surface MICA/B was analyzed by flow cytometry. (F) Effects of romidepsin and 7C6 in CD45+ CD33+ CD34+ primary cells from de novo AML patients. Peripheral blood mononuclear cells from patients with AML were treated for 24 hours with the indicated doses of romidepsin plus 20 μg/mL of the indicated antibodies. Subsequently, the surface MICA/B levels were analyzed by flow cytometry with antibody panel (CD45, CD33, CD34, and MICA/B) plus Zombie Yellow. Romidepsin and 7C6 increase the surface MICA/B expression levels in AML patient-derived cells. Data are representative of (A-B,D-E) 3 independent experiments, and are (B,D-F) mean ± SD of triplicates, except for panel E LSC 0 nmol/L romidepsin isotype that are duplicates. The P values were calculated with 2-way ANOVA. ***P < .001 (calculated with Bonferroni’s post hoc test).
An HDAC 1 and 2-specific inhibitor, romidepsin, synergizes with 7C6 to increase the surface MICA/B expression levels. (A) Romidepsin increases MICB mRNA expression. NB4 cells were treated for 24 hours with 10 nmol/L of the indicated inhibitors or equal volume of dimethyl dioxide. Subsequently, mRNA was extracted, cDNA was synthetized, and PCR was performed with primers specific for the indicated genes plus GAPDH. Analyses were performed via electrophoresis and ImageJ software with GAPDH normalization. (B-C) Romidepsin increases MICB expression for stabilization of the surface protein by 7C6. NB4 cells were treated for 24 hours with 20 μg/mL of the indicated antibodies plus the indicated concentrations of romidepsin. Subsequently, soluble MICA/B shed into culture supernatants were analyzed by sandwich ELISA kits specific for either MICA or MICB. Surface MICA/B and cellular viability were analyzed by flow cytometry upon labeling of cells with 6D4 and Zombie Yellow, respectively. (D) Romidepsin + 7C6 treatment of leukemia cells enhances phagocytosis. NB4 cells were treated for 24 hours with romidepsin or equal volume of phosphate-buffered saline + 20 μg/mL of the indicated antibodies, and subsequently labeled with CFSE. Monocyte-derived macrophages from healthy donors were cocultured for 1 hour with the treated NB4 cells, and phagocytosis was analyzed by flow cytometry via identification of CD14+ CFSE+ macrophages. (E) Romidepsin and 7C6 increase the surface MICA/B expression levels in iPSC-derived AML blasts and LSC. The indicated cells were treated for 24 hours with the indicated concentrations of romidepsin plus 20 μg/mL of the indicated antibodies, and surface MICA/B was analyzed by flow cytometry. (F) Effects of romidepsin and 7C6 in CD45+ CD33+ CD34+ primary cells from de novo AML patients. Peripheral blood mononuclear cells from patients with AML were treated for 24 hours with the indicated doses of romidepsin plus 20 μg/mL of the indicated antibodies. Subsequently, the surface MICA/B levels were analyzed by flow cytometry with antibody panel (CD45, CD33, CD34, and MICA/B) plus Zombie Yellow. Romidepsin and 7C6 increase the surface MICA/B expression levels in AML patient-derived cells. Data are representative of (A-B,D-E) 3 independent experiments, and are (B,D-F) mean ± SD of triplicates, except for panel E LSC 0 nmol/L romidepsin isotype that are duplicates. The P values were calculated with 2-way ANOVA. ***P < .001 (calculated with Bonferroni’s post hoc test).
We cotreated NB4 cells with 7C6 (with a human IgG1, hIgG1) or isotype plus romidepsin. Multiparameter analyses revealed that romidepsin lowers the cellular viability after 24 hours, and increases the levels of soluble MICB shed into culture supernatants but to lower extent in the supernatants from 7C6-hIgG1-treated NB4. Romidepsin also increased the surface MICA/B levels, as determined by staining with 6D4 antibody that does not discriminate MICA from MICB. The 7C6-hIgG1 approximately doubled the mean levels of surface MICA/B on romidepsin-treated NB4 (Figure 4B-C); these cells were also phagocytosed by human monocyte-derived macrophages at higher levels compared with control NB4 (Figure 4D). iPSCs generated from a patient with AML can produce LSCs that differentiate to AML blasts.19 The 7C6-hIgG1 plus romidepsin increased surface MICA/B in iPSC-derived AML blasts and LSC (Figure 4E; supplemental Figure 10). We confirmed these effects with peripheral blood mononuclear cells from 3 untreated de novo AML patients, by identifying the leukemia cells with antibody panel (supplemental Figure 11A-C). We used 7C6-DANA to minimize the Fc receptor-mediated destruction of leukemia cells. In a dose-dependent manner, romidepsin synergized with 7C6-DANA to increase surface MICA/B in these primary leukemia cells (Figure 4F; supplemental Figure 11D). Therefore, romidepsin and 7C6 increase the expression of MICA/B in human AML.
7C6 + romidepsin inhibits human AML in vivo
NB4 engrafts into NSG mice (Figure 5A; supplemental Figure 12A). Twenty-three days after inoculation, these mice were treated twice, separately on 2 subsequent days, with the romidepsin doses of 0, 2.5, 5, and 10 mg/kg plus 7C6-DANA administered by IP injections. Because NB4 does not express MICA/B in vitro (Figure 4B), we omitted the isotype treatment groups to minimize the numbers of mice needed to accomplish this experiment’s goal, which was to increase NB4’s surface MICA/B in vivo. Upon submandibular bleedings of a subset of mice, we confirmed that blood NB4 does not express surface MICA/B before treatment. The 7C6-DANA increased surface MICA/B in blood NB4 isolated from romidepsin-treated mice (Figure 5B; supplemental Figure 12B). However, romidepsin did not increase MICA/B in NB4 cells that were in the bone marrow (Figure 5C; supplemental Figure 12C).

The 7C6 + romidepsin combination therapy increases MICA/B in vivo and inhibits AML. (A-E) NSG mice were inoculated IV with 2 × 106 NB4 cells, which were then analyzed ∼3 weeks later by flow cytometry for identification of human CD45+ human CD33+ mouse CD45.1− cells in the bloods and femoral bone marrows. (A) A drawing and representative flow cytometry plots illustrating this human AML model. (B-C) In 7C6-treated mice, romidepsin enhances the surface MICA/B levels in NB4 cells (B) from the blood (C) but not bone marrow. On day 23 after NB4 inoculation, a subset of mice was bled via submandibular bleeding to analyze the surface MICA/B levels on NB4 by flow cytometry. Subsequently, the mice were treated with the indicated doses of romidepsin plus 0.2 mg 7C6-hIgG1-DANA on days 23 and 24. On day 25, mice were euthanized via CO2 and the surface MICA/B levels on NB4 cells isolated from the (B) blood, and (C) femoral bone marrows were analyzed by flow cytometry. (D-E) Combination therapy with romidepsin and 7C6. On days 18 and 19 after NB4 inoculation, mice were treated with the indicated doses of romidepsin + 0.2 mg of the indicated antibodies. Analysis of leukemia cells was done 1 week later by flow cytometry. Analyses of NB4 cells in the (D) blood and (E) femoral bone marrow are shown. (B-E) Data are median ± interquartile range and are pooled of 2 independent experiments. *P < .05. **P < .01 (Mann-Whitney tests comparing 2 groups at a time).
The 7C6 + romidepsin combination therapy increases MICA/B in vivo and inhibits AML. (A-E) NSG mice were inoculated IV with 2 × 106 NB4 cells, which were then analyzed ∼3 weeks later by flow cytometry for identification of human CD45+ human CD33+ mouse CD45.1− cells in the bloods and femoral bone marrows. (A) A drawing and representative flow cytometry plots illustrating this human AML model. (B-C) In 7C6-treated mice, romidepsin enhances the surface MICA/B levels in NB4 cells (B) from the blood (C) but not bone marrow. On day 23 after NB4 inoculation, a subset of mice was bled via submandibular bleeding to analyze the surface MICA/B levels on NB4 by flow cytometry. Subsequently, the mice were treated with the indicated doses of romidepsin plus 0.2 mg 7C6-hIgG1-DANA on days 23 and 24. On day 25, mice were euthanized via CO2 and the surface MICA/B levels on NB4 cells isolated from the (B) blood, and (C) femoral bone marrows were analyzed by flow cytometry. (D-E) Combination therapy with romidepsin and 7C6. On days 18 and 19 after NB4 inoculation, mice were treated with the indicated doses of romidepsin + 0.2 mg of the indicated antibodies. Analysis of leukemia cells was done 1 week later by flow cytometry. Analyses of NB4 cells in the (D) blood and (E) femoral bone marrow are shown. (B-E) Data are median ± interquartile range and are pooled of 2 independent experiments. *P < .05. **P < .01 (Mann-Whitney tests comparing 2 groups at a time).
We repeated this human AML model with 7C6-mIgG2a to enable phagocytosis by murine macrophages. However, a caveat is that the NOD mouse strain, which is the background of NSG mice, has a polymorphism in Sirpa that enhances binding to human CD47. As consequence, NOD mice enable better engraftment of human cells because macrophages perform less phagocytosis.41 Treatments with 7C6-mIgG2a plus 2.5 mg/kg romidepsin lowered the frequency and absolute number of NB4 in the blood of NSG mice, compared with the mice treated with isotype plus 2.5 mg/kg romidepsin. There was an apparent decrease in the leukemia burden in mice treated with 7C6-mIgG2a plus 5 mg/kg romidepsin compared with isotype plus 5 mg/kg romidepsin, but this difference did not reach statistical significance. Furthermore, 7C6-mIgG2a plus romidepsin at 2.5 or 5 mg/kg apparently lowered the leukemia burden if compared with 7C6-mIgG2a without romidepsin, but this was again not statistically significance (Figure 5D). Interestingly, in the bone marrow, romidepsin alone lowered the frequency and absolute numbers of NB4 cells; this effect was independent of 7C6 (Figure 5E). Collectively, these data indicate that romidepsin plus 7C6 increases the expression of MICA/B by human leukemia cells in vivo and lowers the human leukemia burden.
Discussion
Here, we showed a mAb that inhibits the MICA/B shedding can also promote macrophage-driven immunity against AML. NK cell depletion did not abrogate the efficacy of MICA/B mAb against AML, but this result does not undermine the immunotherapeutic potential of NK cells. In a previous study with melanoma models, we found that MHC class I on tumor cells inhibits the therapeutic efficacy of 7C6 by engaging NK cell inhibitory receptors.24 C1498 expresses MHC class I.42 As such, NK cells may be suppressed by inhibitory receptors that are not neutralized by 7C6 in this AML model. C1498 also expresses an endogenous NKG2D ligand (MULT-1) that may compensate the downregulation of MICA/B, thus enabling NKG2D recognition even in absence of 7C6. In agreement with these possibilities, here we discovered that 7C6 inhibits AML primarily via Fc-receptor signaling that triggers antibody-dependent phagocytosis of leukemia cells by macrophages. In this setting, the MICA/B cellular stress markers serve as leukemia antigens. However, the MICA/B antibody’s mechanism of action against AML appears to involve multiple leukocyte populations and be dependent on the tissue microenvironment: primarily macrophage mediated in the blood, whereas macrophage, CD8 T cell, and NKT cell mediated in the bone marrow. It is possible that CD8 T cells and NKT cells provide cytokine-mediated support to macrophages in the bone marrow, such as via interferon-γ. Finally, the tumor model could also play a role because in melanoma models we found that 7C6 inhibits metastases primarily by promoting NK cell-driven immunity.18,24
Approximately one-half of patients with AML lack soluble MICA/B in the plasma.34 LSCs, which are progenitor cells with self-renewal capacity and that drive disease relapse, also do not express MICA/B.35 If tested in clinical trials, 7C6 would be selective for MICA/B+ AML. We bypassed this limitation with romidepsin, a selective HDAC inhibitor approved for cutaneous T-cell lymphoma, and caused only manageable toxicities in patients with AML in 2 previous trials.43,44 However, HDAC inhibitors in general have not generated response rates and allowed AML progression.45 Here, romidepsin was assigned to a new task: increase MICA/B mRNA expression for surface protein stabilization by 7C6 and subsequent immune reaction. In the clinical trials with patients with AML, which had no 7C6, the ability of HDAC inhibitors in increasing MICA/B expression would not have had an impact because of cleavage.43-45 Romidepsin is also apparently more potent than panobinostat and mocetinostat, although their selectivities overlap because romidepsin increased MICB mRNA expression at a low dose where other inhibitors were ineffective. Interestingly, although we found that MICA/B expression was increased only in leukemia cells circulating in the peripheral blood, romidepsin lowered the leukemia burden in the bone marrow compartment. This indicates that bone marrow leukemia cells are targeted by romidepsin, but signals from the bone marrow microenvironment suppress MICA/B expression. Therefore, the combination therapy stimulates immunity against circulating leukemia cells, whereas in the bone marrow the therapeutic effects are likely driven by romidepsin’s direct cytotoxic actions.
A limitation in our study is that NSG mice have the NOD mouse background, which has a variant of the SIRPA protein that binds with high affinity to human CD47.41 The SIRPA–CD47 axis represents a “do not eat me” signal that inhibits macrophages, and CD47 is upregulated by AML cells.32 Another limitation is that the murine AML cell lines express 2 foreign antigens (MICB and ZsGreen), which have the potential of inducing a T cell-driven immunity in mice but that may not be induced in patients.
In summary, we showed that 7C6 inhibits the AML outgrowth via antibody-dependent phagocytosis and romidepsin upregulates MICA/B for subsequent binding by 7C6, which is followed by phagocytosis. These results provide the evidence-based rationale for transition to clinical phases. Therefore, our approach helps diversify the immunotherapeutic armamentarium available for future clinical trials with AML patients.
Acknowledgments
The authors thank Sergio Lira, Ramon Parsons, Brian Brown, and Miriam Merad for helpful discussions; Ronald Hoffman and Bridget Marcellino for the primary acute myeloid leukemia culture protocol and also helpful discussions; Siraj El Jamal for helping with histopathology of the hematoxylin and eosin slides; and the Hematological Malignancies Tissue Bank at Mount Sinai for providing the patient samples.
L.F.d.A. is the recipient of a Cancer Research Institute Clinic and Laboratory Integration Program (CLIP) Grant (CRI Award #CRI3483).
Authorship
Contribution: P.H.A.d.S. performed several of the MICA/B shedding and phagocytosis assays, produced recombinant 7C6 antibody, and did quality of control tests; S.X. also performed several of the MICA/B shedding and phagocytosis assays; A.G.K. and E.P.P. produced the iPSC-derived AML blasts; L.S.C. and X.S. supervised the statistical analyses; K.W.W. and J.M. helped to conceive the study and write the article; K.W.W. also provided CHO cells that produce the recombinant 7C6 antibodies; and L.F.d.A. conceived the study, performed all the in vivo experiments, and wrote the manuscript.
Conflict-of-interest disclosure: L.F.d.A. and K.W.W. are inventors of published patents (PCT/US2018/033793 and PCT/US2020/055009). L.F.d.A. also served as a consultant to Cullinan Oncology. The remaining authors declare no competing financial interests.
Correspondence: Lucas Ferrari de Andrade, Icahn School of Medicine at Mount Sinai, 1425 Madison Ave, Suite 12-20B, PO Box 1630, New York, NY 10029; e-mail: lucas.ferrarideandrade@mssm.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal