

Key Points
Ibrutinib is effective in patients with early-stage CLL, but the results do not justify changing the current standard of “watch and wait.”
Ibrutinib is associated with relevant cardiovascular toxicity.

Abstract
Observation is the current standard of care for patients with early-stage asymptomatic chronic lymphocytic leukemia (CLL), as chemotherapy-based interventions have failed to prolong survival. We hypothesized that early intervention with ibrutinib would be well tolerated and lead to superior disease control in a subgroup of early-stage patients with CLL. The phase 3, double-blind, placebo-controlled CLL12 trial randomly assigned asymptomatic, treatment-naïve Binet stage A CLL patients at increased risk of progression in a 1:1 ratio to receive ibrutinib (n = 182) or placebo (n = 181) at a dose of 420 mg daily. At a median follow-up of 31 months, the study met its primary endpoint by significantly improving event-free survival in the ibrutinib group (median, not reached vs 47.8 months; hazard ratio = 0.25; 95% confidence interval = 0.14-0.43, P < .0001). Compared with placebo, ibrutinib did not increase overall toxicity, yielding similar incidence and severity of adverse events (AEs). The most common serious AEs were atrial fibrillation, pneumonia, and rash in the ibrutinib group, and basal cell carcinoma, pneumonia, and myocardial infarction in the placebo group. Ibrutinib-associated risk for bleeding (33.5%) was decreased by prohibiting the use of oral anticoagulants through an amendment of the study protocol and by avoiding CYP3A4 drug–drug interactions. Ibrutinib confirms efficacy in CLL patients at an early stage with an increased risk of progression. However, the results do not justify changing the current standard of “watch and wait.” This trial was registered at www.clinicaltrials.gov as #NCT02863718.
Introduction
The majority of patients with chronic lymphocytic leukemia (CLL) are diagnosed at an early asymptomatic stage. The current standard is a “watch and wait” strategy until clinical signs of progressive bone marrow failure or symptoms caused by leukemia are observed.1 In recent years, several targeted drugs have been approved for CLL treatment, showing high efficacy and lower toxicity than chemotherapy.2-6 Ibrutinib inhibits the Bruton’s tyrosine kinase (BTK) and has shown sustained benefits compared with standard chemoimmunotherapy in treatment-naïve and relapsed CLL.2-4,7,8
The clinical staging systems introduced by Rai et al and Binet et al have been the backbone of prognostication in CLL.9,10 Advanced scoring systems have integrated genetic and biological parameters to improve the prediction of the clinical course, in particular at early stages.11-14 A score of the German CLL study group proposed 4 patient categories of different overall survival (OS).13 This approach was later adopted by a consortium of international investigators to create the international prognostic index for CLL (CLL-IPI).11 Using these scores, CLL patients with an elevated risk for rapid progression and an early need of treatment can be identified.
In this context, we wished to evaluate whether early-stage CLL patients with increased risk of progression would benefit from early ibrutinib treatment. For this purpose, we designed the CLL12 trial, a placebo-controlled, double-blind, randomized trial including early-stage (Binet A) CLL patients at increased risk of progression as identified by the German CLL study group score. This is the first trial comparing targeted agents in early-stage CLL to placebo. Here we report the results of the planned, primary endpoint analysis.
Methods
Patients
Eligible patients were required to have a centrally confirmed diagnosis of CLL in Binet stage A without treatment indication, were at least 18 years of age, had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0 to 2, and a life expectancy of at least 6 months. The inclusion criteria did not contain restrictions on upper age nor the cumulative illness rating scale score.15 Exclusion criteria included any prior CLL-specific therapy, impaired renal or liver function, and concomitant anticoagulant treatment with warfarin, phenprocoumon, or other vitamin K-antagonists as well as strong CYP3A4 inhibitors. The protocol was amended on 15 May 2015 to exclude patients with concomitant use of direct Xa inhibitors (supplemental File, available on the Blood Web site).
Study design and treatment
CLL12 is an investigator-initiated, phase 3, randomized, multicenter, placebo-controlled, double-blinded trial performed at 89 academic and community hospitals and oncology practices in Germany.
Risk of disease progression was measured in all patients using the German CLL study group (GCLLSG) score by 8 differently weighted factors (1-6 points): age >60 years (1), male sex (1), β2-microglobulin 1.7 to 3.5 mg/L (1) or >3.5 mg/L (2), ECOG performance status >0 (1), thymidine kinase >10 U/L (2), unmutated immunoglobulin heavy chain gene (IGHV) status (1), 11q deletion (1) and 17p deletion (6) (supplemental Table 1).13 The 4 different risk categories produced by the GCLLSG score (GCLLSG risk groups) were used to allocate patients to 3 different arms: low-risk patients (score 0-2) were allocated to the observational cohort; intermediate (score 3-5), high (score 6-10), and very high-risk patients (score >10) were randomized in a 1:1 ratio to receive ibrutinib or placebo. Randomization was stratified according to GCLLSG risk groups and the presence of TP53 mutations.
Patients, site staff, and the sponsor’s clinical and medical representatives were unaware of treatment group assignments. Ibrutinib and placebo capsules were identical in appearance. The blinding was not broken throughout the study unless it was absolutely considered essential. Patients were instructed to take 3 capsules (for a dose of 420 mg) once daily. One cycle was defined as 28 calendar days. Dose modifications were allowed for the management of adverse events (AEs). Study drug could be paused for a maximum of 28 consecutive days.
Staging procedures included a complete blood count and a physical examination. Response was evaluated every 3 cycles according to the 2008 International Workshop on CLL (iwCLL) criteria.16 Treatment-related lymphocytosis within the first 6 cycles was not considered progressive disease in the absence of other iwCLL criteria for progression. AEs were graded according to the NCI Common Toxicity Criteria.
The trial was approved by the institutional review board at each participating institution and was conducted in accordance with the Declaration of Helsinki and International Conference on Harmonization Guidelines for Good Clinical Practice. All patients provided written informed consent. An independent data-monitoring committee periodically reviewed unblinded safety data and efficacy results of the primary endpoint analysis.
Study endpoints and assessments
The primary endpoint was event-free survival (EFS), defined as the time from randomization to active disease progression with treatment indication according to the iwCLL criteria or initiation of subsequent treatment of CLL or death from any cause.16 Secondary endpoints for treatment arms included progression-free survival (PFS), time to next CLL treatment, treatment-free survival (TFS), OS, best overall response rate achieved during treatment or within 6 months after the end of treatment, duration of response, response to subsequent treatment, and safety. As defined per protocol, TFS and OS were not analyzed for the primary endpoint analysis.
Statistical analysis
Sample size regarding treatment groups was estimated on the basis of an assumed hazard ratio (HR) of 0.5 for active disease progression, initiation of subsequent treatment, or death, with 71 events providing a power of 80% on the basis of a 2-sided log-rank test stratified according to GCLLSG risk groups, with an α level of 0.05. These estimates assumed a median EFS of 48 months in the ibrutinib group compared with 24 months in the placebo group (based on unpublished external data of the GCLLSG CLL1, CLL4, and CLL8 study for Binet stage A CLL patients) (supplemental Figure 1).13 The CLL12 trial met its primary endpoint at the data cutoff of 7 March 2019.
During conduct of the study, it was decided to enable hypothesis testing for statistical superiority regarding the secondary endpoint OS. A total of 47 survival events are required to detect an HR of 0.4 with 80% power in a 2-sided log-rank test with a significance level of 5% (adjusted for 1 interim analysis with 28 survival events [60%] and stratified according to GCLLSG risk groups). These estimates corresponded to a median OS of 120 months in the placebo group and assumed a prolongation to 300 months in the ibrutinib-treated group.
Efficacy analyses were based on the intention-to-treat principle, including all patients who underwent randomization to treatment arms (ie, patients with intermediate, high, or very high GCLLSG risk [treatment cohort]) (Figure 1). The sensitivity analysis of EFS was performed in the per-protocol set, which comprised all patients who had completed at least 2 cycles of study treatment unless they progressed or died before, provided they fulfilled the inclusion criteria and had no major protocol violations (supplemental Table 11). All patients receiving any amount of study drug were considered evaluable for safety analyses (safety cohort) (Figure 1). To control for type I error, a hierarchical sequential testing procedure was considered for secondary time-to-event endpoints, PFS, and time to next CLL treatment (using log-rank test stratified by GCLLSG risk groups).17
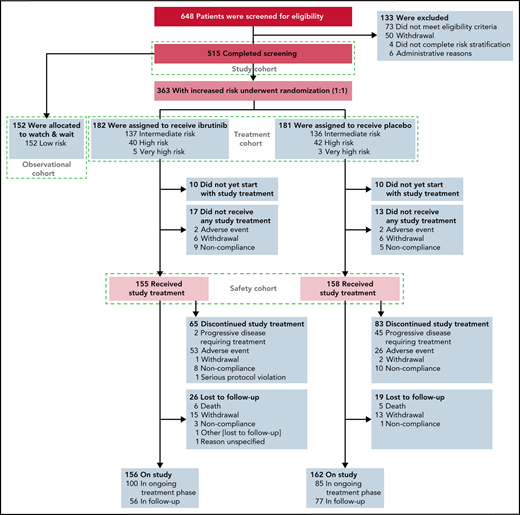
Screening, risk stratification, randomization, and follow-up in the phase 3 CLL12 study. The 363 patients with increased risk of disease progression were randomly assigned to a treatment arm (n = 182 in the ibrutinib group and n = 181 in the placebo group).
Screening, risk stratification, randomization, and follow-up in the phase 3 CLL12 study. The 363 patients with increased risk of disease progression were randomly assigned to a treatment arm (n = 182 in the ibrutinib group and n = 181 in the placebo group).
The “watch and wait” cohort (observation cohort) (Figure 1) included all patients assessed to be low GCLLSG risk and was not part of the primary endpoint analysis.
Results
Patient characteristics and treatment
From 5 May 2014 to 14 February 2019, 648 patients consented to participate in the study and were screened for eligibility, of which 133 patients were excluded. A total of 515 patients were enrolled, 152 low-risk patients were allocated to the observational cohort, 182 and 181 patients were randomly assigned to receive ibrutinib and placebo (treatment cohort), respectively (Figure 1). The GCLLSG risk was intermediate in 273 (75.2%) of the 363 randomized patients, high risk in 82 (22.6%), and very high risk in 8 (2.2%) (supplemental Table 2).13 The demographic and disease characteristics were well balanced between the 2 treatment groups (Table 1). Median age was 64 years; 140 (38.7%) patients had unmutated IGHV genes, 13 (3.6%) had chromosome del(17p), and 27 (7.4%) patients had TP53 mutations. Median time from the first diagnosis to randomization was 7.9 months (range, 0.7-222.4; interquartile range [IQR], 3.4-35.7) and 11.5 months (range, 0.7-285.3; IQR, 2.8-35.7) for the ibrutinib and placebo group, respectively.
Selected baseline characteristics of the patients (treatment cohort)*
| Characteristics | Ibrutinib group (n = 182) | Placebo group (n = 181) | Total (n = 363) |
|---|---|---|---|
| Age, y | |||
| Median | 64 | 64 | 64 |
| Range | 38-85 | 36-80 | 36-85 |
| >60, n (%) | 114 (62.6) | 115 (63.5) | 229 (63.1) |
| >70, n (%) | 40 (22.0) | 41 (22.7) | 81 (22.3) |
| Male sex, n (%) | 137 (75.3) | 127 (70.2) | 264 (72.7) |
| Time between first diagnosis and randomization, mo | 182 | 179 | 361 |
| Median | 7.9 | 11.5 | 9.3 |
| Mean | 25.1 | 27.1 | 26.1 |
| Standard deviation | 33.9 | 39.2 | 36.6 |
| Interquartile range | 3.4-35.7 | 2.8-35.7 | 3.1-35.7 |
| Range | 0.7-222.4 | 0.7-285.3 | 0.7-285.3 |
| ECOG performance status, n (%)† | |||
| 0 | 163 (89.6) | 160 (88.4) | 323 (89.0) |
| 1 | 19 (10.4) | 21 (11.6) | 40 (11.0) |
| β2 microglobulin (mg/L) | |||
| ≤1.7 | 10 (5.5) | 16 (8.8) | 26 (7.2) |
| >1.7 ≤ 3.5 | 158 (86.8) | 151 (83.4) | 309 (85.1) |
| >3.5 | 14 (7.7) | 14 (7.7) | 28 (7.7) |
| Thymidine kinase (U/L) | |||
| >10 | 138 (75.8) | 150 (82.9) | 288 (79.3) |
| Cytogenetic subgroup according to hierarchical Döhner classification, n (%)‡ | |||
| Deletion in 17p | 6 (3.3) | 7 (3.9) | 13 (3.6) |
| Deletion in 11q | 21 (11.5) | 19 (10.5) | 40 (11.0) |
| Trisomy in 12 | 24 (13.2) | 28 (15.5) | 52 (14.3) |
| No abnormalities | 36 (19.8) | 30 (16.6) | 66 (18.2) |
| Deletion in 13q alone | 95 (52.2) | 97 (53.6) | 192 (52.9) |
| Mutated TP53, n (%) | 14 (7.7) | 13 (7.2) | 27 (7.4) |
| IGHV mutation status, n (%)§ | 181 | 181 | 362 |
| Mutated | 111 (61.3) | 109 (60.2) | 220 (60.8) |
| Unmutated | 70 (38.7) | 70 (38.7) | 140 (38.7) |
| Not evaluable | 0 (0.0) | 2 (1.1) | 2 (0.6) |
| GCLLSG risk group, n (%) | |||
| Intermediate | 137 (75.3) | 136 (75.1) | 273 (75.2) |
| High | 40 (22.0) | 42 (23.2) | 82 (22.6) |
| Very high | 5 (2.7) | 3 (1.7) | 8 (2.2) |
| CLL-IPI risk group, n (%) | 181 | 181 | 362 |
| Low | 98 (54.1) | 98 (54.1) | 196 (54.1) |
| Intermediate | 59 (32.6) | 58 (32.0) | 117 (32.3) |
| High | 22 (12.2) | 23 (12.7) | 45 (12.4) |
| Very high | 2 (1.1) | 2 (1.1) | 4 (1.1) |
| Characteristics | Ibrutinib group (n = 182) | Placebo group (n = 181) | Total (n = 363) |
|---|---|---|---|
| Age, y | |||
| Median | 64 | 64 | 64 |
| Range | 38-85 | 36-80 | 36-85 |
| >60, n (%) | 114 (62.6) | 115 (63.5) | 229 (63.1) |
| >70, n (%) | 40 (22.0) | 41 (22.7) | 81 (22.3) |
| Male sex, n (%) | 137 (75.3) | 127 (70.2) | 264 (72.7) |
| Time between first diagnosis and randomization, mo | 182 | 179 | 361 |
| Median | 7.9 | 11.5 | 9.3 |
| Mean | 25.1 | 27.1 | 26.1 |
| Standard deviation | 33.9 | 39.2 | 36.6 |
| Interquartile range | 3.4-35.7 | 2.8-35.7 | 3.1-35.7 |
| Range | 0.7-222.4 | 0.7-285.3 | 0.7-285.3 |
| ECOG performance status, n (%)† | |||
| 0 | 163 (89.6) | 160 (88.4) | 323 (89.0) |
| 1 | 19 (10.4) | 21 (11.6) | 40 (11.0) |
| β2 microglobulin (mg/L) | |||
| ≤1.7 | 10 (5.5) | 16 (8.8) | 26 (7.2) |
| >1.7 ≤ 3.5 | 158 (86.8) | 151 (83.4) | 309 (85.1) |
| >3.5 | 14 (7.7) | 14 (7.7) | 28 (7.7) |
| Thymidine kinase (U/L) | |||
| >10 | 138 (75.8) | 150 (82.9) | 288 (79.3) |
| Cytogenetic subgroup according to hierarchical Döhner classification, n (%)‡ | |||
| Deletion in 17p | 6 (3.3) | 7 (3.9) | 13 (3.6) |
| Deletion in 11q | 21 (11.5) | 19 (10.5) | 40 (11.0) |
| Trisomy in 12 | 24 (13.2) | 28 (15.5) | 52 (14.3) |
| No abnormalities | 36 (19.8) | 30 (16.6) | 66 (18.2) |
| Deletion in 13q alone | 95 (52.2) | 97 (53.6) | 192 (52.9) |
| Mutated TP53, n (%) | 14 (7.7) | 13 (7.2) | 27 (7.4) |
| IGHV mutation status, n (%)§ | 181 | 181 | 362 |
| Mutated | 111 (61.3) | 109 (60.2) | 220 (60.8) |
| Unmutated | 70 (38.7) | 70 (38.7) | 140 (38.7) |
| Not evaluable | 0 (0.0) | 2 (1.1) | 2 (0.6) |
| GCLLSG risk group, n (%) | |||
| Intermediate | 137 (75.3) | 136 (75.1) | 273 (75.2) |
| High | 40 (22.0) | 42 (23.2) | 82 (22.6) |
| Very high | 5 (2.7) | 3 (1.7) | 8 (2.2) |
| CLL-IPI risk group, n (%) | 181 | 181 | 362 |
| Low | 98 (54.1) | 98 (54.1) | 196 (54.1) |
| Intermediate | 59 (32.6) | 58 (32.0) | 117 (32.3) |
| High | 22 (12.2) | 23 (12.7) | 45 (12.4) |
| Very high | 2 (1.1) | 2 (1.1) | 4 (1.1) |
Percentages may not total 100 because of rounding. All but 1 patient of each group had Binet stage A at baseline.
ECOG performance-status scores are assessed on a 5-point scale, with higher scores indicating greater disability.
Cytogenetic subgroups were determined according to the hierarchical model of Döhner et al.31
IGHV mutation status was tested in 362 patients and could be determined in 360 patients.
At the data cutoff for primary endpoint analysis, 100 patients of the ibrutinib group (54.9%) were still receiving treatment compared with 85 patients of the placebo group (47.0%). Sixty-five patients of the ibrutinib (35.7%) and 83 patients of the placebo group (45.9%) discontinued treatment early (Figure 1). The most frequent reasons for treatment discontinuation were AEs in the ibrutinib group (81.5%) and disease progression in the placebo group (54.2%). The majority (57.1%) of treatment discontinuations caused by AEs occurred within the first 6 treatment cycles.
The median number of treatment cycles was 21 (range, 1-57) with a median treatment duration of 18.4 months (range, 0.1-52.4) for ibrutinib and 18 cycles (range, 1-57) with a median treatment duration of 16.3 months (range, 0.1-51.5) for the placebo group. Among the 65 patients who discontinued ibrutinib prematurely, the median treatment duration was 5.5 months (range, 0.1-52.4). Dose reductions of at least 20% of the planned dose per cycle were documented in 16.6% cycles of ibrutinib and 2.5% cycles of placebo. The most common reason for dose reduction was AEs affecting 14.2% and 1.8% cycles in the ibrutinib and placebo groups, respectively.
Efficacy
With a median observation time of 31.0 months (range, 0-56.3), significantly fewer patients had a primary endpoint event in the ibrutinib group (18 of 182 patients [9.9%]) than in the placebo group (55 of 181 patients [30.4%]). The HR for EFS (as defined by progression to active disease, initiation of subsequent CLL treatment, or death) was 0.25 (95% confidence interval [CI], 0.14-0.43; P < .0001 by stratified log-rank test) (Figure 2). The 3-year EFS rate was 87.3% (95% CI, 81.3% to 93.2%) in the ibrutinib group and 60.4% (95% CI, 51.4% to 69.5%) in the placebo group. This treatment effect of ibrutinib was observed across all major subgroups (Figure 3).
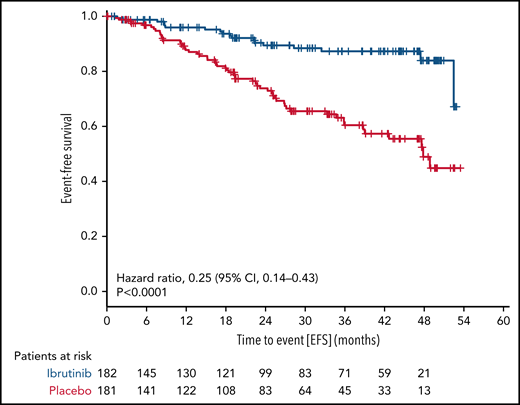
EFS. The primary endpoint of EFS is shown for all patients by therapy received.
EFS. The primary endpoint of EFS is shown for all patients by therapy received.
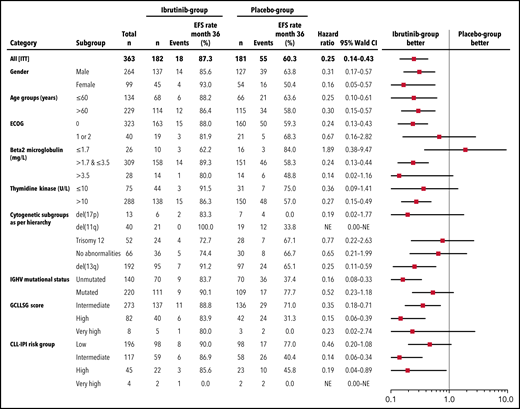
Subgroup analysis of EFS. Forest plot showing EFS by prespecified subgroups according to baseline demographic and clinical characteristics.
Subgroup analysis of EFS. Forest plot showing EFS by prespecified subgroups according to baseline demographic and clinical characteristics.
In the per-protocol set, 13 of 136 patients (9.6%) in the ibrutinib group and 51 of 149 patients (34.2%) in the placebo group had primary endpoint events (HR, 0.19; 95% CI, 0.10-0.35; 3-year EFS rate, 89.5% [95% CI, 83.5% to 95.5%] vs 60.1% [95% CI, 50.7% to 69.5%], respectively) (supplemental Figure 4; supplemental Table 11).
The median PFS (including asymptomatic disease progressions not requiring treatment) was not reached in the ibrutinib group (30 events of progression or death in 182 patients [16.5%]) and was 14.8 months in the placebo group (101 events in 181 patients [55.8%]; HR for progression or death, 0.18 [95% CI, 0.12-0.27; P < .0001 by stratified log-rank test) (Figure 4A). The 3-year PFS rate was higher in the ibrutinib group than in the placebo group (80.9% [95% CI, 74.0% to 87.7%] vs 28.5% [95% CI, 20.5% to 36.5%]).
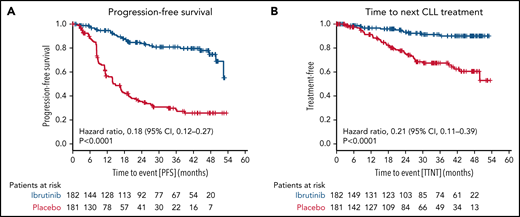
Secondary endpoints. The secondary endpoints are shown for all patients by therapy received for PFS (A) and time to next CLL treatment (B).
Secondary endpoints. The secondary endpoints are shown for all patients by therapy received for PFS (A) and time to next CLL treatment (B).
Time to next CLL treatment was longer in the ibrutinib group (Kaplan-Meier estimate of percentage of patients without initiation of next CLL treatment at 36 months, 91.3% [95% CI, 86.2% to 96.3%] vs 67.5% [95% CI, 59.2% to 75.7%], respectively; HR for initiation of next CLL treatment 0.21 [95% CI, 0.11-0.39]; P < .0001 by stratified log-rank test) (Figure 4B).
In the ibrutinib group, 120 of 182 (65.9%) patients responded to treatment, and the median duration of response was not reached. In 25 (13.7%) patients, response data were not yet available, and in 37 (20.3%) patients, stable disease was noted at the time of the data cutoff.
Subsequent treatment was administered to 12 patients of the ibrutinib group (6.6%) as compared with 47 patients in the placebo group (26.0%) (supplemental Tables 9 and 10).
Safety
Three hundred thirteen patients were included in the safety analysis. Three patients of the placebo group received ibrutinib due to incorrect study drug release at the treating center and were allocated to the ibrutinib safety cohort.
Overall, 150 of 158 subjects (94.9%) in the ibrutinib group and 147 of 155 subjects (94.8%) in the placebo group had at least 1 AE. The most common preferred terms with an incidence of at least 20% were nasopharyngitis and fatigue for subjects of both groups, as well as diarrhea for subjects treated with ibrutinib (Table 2). Infections of any CTC grade were reported in 101 (63.9%, 77 [48.7%] grade 1-2) and in 110 (71%, 88 [56.8%] grade 1-2) of patients treated with ibrutinib and placebo, respectively.
Adverse events (safety cohort)
| Ibrutinib (n = 158) | Placebo (n = 155) | |||||
|---|---|---|---|---|---|---|
| Any grade | Grade 1-2 | Grade ≥3 | Any grade | Grade 1-2 | Grade ≥3 | |
| Total no. of events | 1593 | 1426 | 167 | 1015* | 885 | 129 |
| Any AE, n (%) | 150 (94.9) | 70 (44.3) | 80 (50.6) | 147 (94.8) | 80 (51.6) | 67 (43.2) |
| Most common AEs occurring in ≥0% of patients in any treatment group, n (%)† | ||||||
| Atrial fibrillation | 19 (12.0) | 9 (5.7) | 10 (6.3) | 2 (1.3) | 2 (1.3) | |
| Diarrhea | 50 (31.6) | 48 (30.4) | 2 (1.3) | 28 (18.1) | 27 (17.4) | 1 (0.6) |
| Dyspepsia | 23 (14.6) | 23 (14.6) | 4 (2.6) | 4 (2.6) | ||
| Nausea | 26 (16.5) | 26 (16.5) | 15 (9.7) | 15 (9.7) | ||
| Fatigue | 40 (25.3) | 39 (24.7) | 1 (0.6) | 32 (20.6) | 31 (20.0) | 1 (0.6) |
| Nasopharyngitis | 42 (26.6) | 41 (25.9) | 1 (0.6) | 51 (32.9) | 51 (32.9) | |
| Upper respiratory tract infection | 16 (10.1) | 15 (9.5) | 1 (0.6) | 11 (7.1) | 11 (7.1) | |
| Arthralgia | 19 (12.0) | 18 (11.4) | 1 (0.6) | 14 (9.0) | 13 (8.4) | 1 (0.6) |
| Back pain | 16 (10.1) | 14 (8.9) | 2 (1.3) | 17 (11.0) | 15 (9.7) | 2 (1.3) |
| Muscle spasms | 22 (13.9) | 6 (3.9) | ||||
| Dizziness | 22 (13.9) | 20 (12.7) | 2 (1.3) | 8 (5.2) | 8 (5.2) | |
| Headache | 28 (17.7) | 28 (17.7) | 17 (11.0) | 17 (11.0) | ||
| Rash | 29 (18.4) | 24 (15.2) | 5 (3.2) | 8 (5.2) | 8 (5.2) | |
| Hematoma | 22 (13.9) | 20 (12.7) | 2 (1.3) | 6 (3.9) | 6 (3.9) | |
| Hypertension | 16 (10.1) | 14 (8.8) | 2 (1.3) | 7 (4.5) | 4 (2.6) | 3 (1.9) |
| Ibrutinib (n = 158) | Placebo (n = 155) | |||||
|---|---|---|---|---|---|---|
| Any grade | Grade 1-2 | Grade ≥3 | Any grade | Grade 1-2 | Grade ≥3 | |
| Total no. of events | 1593 | 1426 | 167 | 1015* | 885 | 129 |
| Any AE, n (%) | 150 (94.9) | 70 (44.3) | 80 (50.6) | 147 (94.8) | 80 (51.6) | 67 (43.2) |
| Most common AEs occurring in ≥0% of patients in any treatment group, n (%)† | ||||||
| Atrial fibrillation | 19 (12.0) | 9 (5.7) | 10 (6.3) | 2 (1.3) | 2 (1.3) | |
| Diarrhea | 50 (31.6) | 48 (30.4) | 2 (1.3) | 28 (18.1) | 27 (17.4) | 1 (0.6) |
| Dyspepsia | 23 (14.6) | 23 (14.6) | 4 (2.6) | 4 (2.6) | ||
| Nausea | 26 (16.5) | 26 (16.5) | 15 (9.7) | 15 (9.7) | ||
| Fatigue | 40 (25.3) | 39 (24.7) | 1 (0.6) | 32 (20.6) | 31 (20.0) | 1 (0.6) |
| Nasopharyngitis | 42 (26.6) | 41 (25.9) | 1 (0.6) | 51 (32.9) | 51 (32.9) | |
| Upper respiratory tract infection | 16 (10.1) | 15 (9.5) | 1 (0.6) | 11 (7.1) | 11 (7.1) | |
| Arthralgia | 19 (12.0) | 18 (11.4) | 1 (0.6) | 14 (9.0) | 13 (8.4) | 1 (0.6) |
| Back pain | 16 (10.1) | 14 (8.9) | 2 (1.3) | 17 (11.0) | 15 (9.7) | 2 (1.3) |
| Muscle spasms | 22 (13.9) | 6 (3.9) | ||||
| Dizziness | 22 (13.9) | 20 (12.7) | 2 (1.3) | 8 (5.2) | 8 (5.2) | |
| Headache | 28 (17.7) | 28 (17.7) | 17 (11.0) | 17 (11.0) | ||
| Rash | 29 (18.4) | 24 (15.2) | 5 (3.2) | 8 (5.2) | 8 (5.2) | |
| Hematoma | 22 (13.9) | 20 (12.7) | 2 (1.3) | 6 (3.9) | 6 (3.9) | |
| Hypertension | 16 (10.1) | 14 (8.8) | 2 (1.3) | 7 (4.5) | 4 (2.6) | 3 (1.9) |
There was 1 serious AE (basal cell carcinoma) with CTC grade missing.
AEs are reported according to the Medical Dictionary for Regulatory Activities preferred terms and National Cancer Institute Common Terminology Criteria for Adverse Events grade.
The incidence of serious AEs was similar in the 2 groups. Serious AEs occurring in at least 2% of the patients were atrial fibrillation and pneumonia in the ibrutinib group and acute myocardial infarction, basal cell carcinoma, and pneumonia in the placebo group (Table 3).
Serious adverse events (safety cohort)
| Ibrutinib (n = 158) | Placebo (n = 155) | |||||
|---|---|---|---|---|---|---|
| Any grade | Grade 1-2 | Grade ≥3 | Any grade | Grade 1-2 | Grade ≥3 | |
| Any serious AEs, n (%) | 62 (39.2) | 11 (6.9) | 51 (32.3) | 58* (37.4) | 4 (2.6) | 53 (34.2) |
| Total n of serious adverse events | 112 | 18 | 94 | 108* | 8 | 99 |
| Serious AEs with ≥2% incidence in either group, n (%)† | ||||||
| Atrial fibrillation | 9 (5.7) | 1 (0.6) | 8 (5.1) | 1 (0.6) | 1 (0.6) | |
| Acute myocardial infarction | 1 (0.6) | 1 (0.6) | 4 (2.6) | 4 (2.6) | ||
| Pneumonia | 7 (4.4) | 7 (4.4) | 6 (3.9) | 6 (3.9) | ||
| Basal cell carcinoma | 3 (1.9) | 1 (0.6) | 2 (1.3) | 6* (3.9) | 5 (3.2) | |
| Any AE of clinical interest, n (%) | 113 (71.5) | 86 (54.4) | 27 (17.1) | 77 (49.7) | 53 (34.2) | 24 (15.5) |
| Total no. of AEs of clinical interest | 280 | 245 | 35 | 164 | 135 | 29 |
| Cardiac arrhythmias | 34 (21.5) | 21 (13.3) | 13 (8.2) | 12 (7.7) | 10 (6.5) | 2 (1.3) |
| Bleeding | 53 (33.5) | 47 (29.7) | 6 (3.8) | 23 (14.8) | 20 (12.9) | 3 (1.9) |
| Hypertensive disorders | 18 (11.4) | 15 (9.5) | 3 (1.9) | 7 (4.5) | 4 (2.6) | 3 (1.9) |
| Cardiac event other than arrhythmia | 12 (7.6) | 7 (4.4) | 5 (3.2) | 16 (10.3) | 7 (4.5) | 9 (5.8) |
| Diarrhea | 63 (39.9) | 60 (38.0) | 3 (1.9) | 46 (29.7) | 39 (25.2) | 7 (4.5) |
| Ibrutinib (n = 158) | Placebo (n = 155) | |||||
|---|---|---|---|---|---|---|
| Any grade | Grade 1-2 | Grade ≥3 | Any grade | Grade 1-2 | Grade ≥3 | |
| Any serious AEs, n (%) | 62 (39.2) | 11 (6.9) | 51 (32.3) | 58* (37.4) | 4 (2.6) | 53 (34.2) |
| Total n of serious adverse events | 112 | 18 | 94 | 108* | 8 | 99 |
| Serious AEs with ≥2% incidence in either group, n (%)† | ||||||
| Atrial fibrillation | 9 (5.7) | 1 (0.6) | 8 (5.1) | 1 (0.6) | 1 (0.6) | |
| Acute myocardial infarction | 1 (0.6) | 1 (0.6) | 4 (2.6) | 4 (2.6) | ||
| Pneumonia | 7 (4.4) | 7 (4.4) | 6 (3.9) | 6 (3.9) | ||
| Basal cell carcinoma | 3 (1.9) | 1 (0.6) | 2 (1.3) | 6* (3.9) | 5 (3.2) | |
| Any AE of clinical interest, n (%) | 113 (71.5) | 86 (54.4) | 27 (17.1) | 77 (49.7) | 53 (34.2) | 24 (15.5) |
| Total no. of AEs of clinical interest | 280 | 245 | 35 | 164 | 135 | 29 |
| Cardiac arrhythmias | 34 (21.5) | 21 (13.3) | 13 (8.2) | 12 (7.7) | 10 (6.5) | 2 (1.3) |
| Bleeding | 53 (33.5) | 47 (29.7) | 6 (3.8) | 23 (14.8) | 20 (12.9) | 3 (1.9) |
| Hypertensive disorders | 18 (11.4) | 15 (9.5) | 3 (1.9) | 7 (4.5) | 4 (2.6) | 3 (1.9) |
| Cardiac event other than arrhythmia | 12 (7.6) | 7 (4.4) | 5 (3.2) | 16 (10.3) | 7 (4.5) | 9 (5.8) |
| Diarrhea | 63 (39.9) | 60 (38.0) | 3 (1.9) | 46 (29.7) | 39 (25.2) | 7 (4.5) |
AEs are reported according to the Medical Dictionary for Regulatory Activities preferred terms and National Cancer Institute Common Terminology Criteria for Adverse Events grade.
AEs of clinical interest include preferred terms, high-level (group) terms and standardized medical queries (SMQ) according to the Medical Dictionary for Regulatory Activities.
The most common grade 3 or higher AEs in the ibrutinib group were atrial fibrillation (10 [6.3%] of 158 patients), pneumonia (7 [4.4%] of 158 patients), and rash (5 [3.2%] of 158 patients), as compared with basal cell carcinoma (8 [5.2%] of 155 patients), pneumonia (7 [4.5%] of 155 patients), and myocardial infarction (5 [3.2%] of 155 patients) in the placebo group.
Special attention was paid to prespecified AEs of interest such as bleeding events, cardiac arrhythmias, hypertensive disorders, cardiac events other than arrhythmia, and diarrhea (Table 2). Bleeding events of any CTC grade (grade 3 or higher) were reported in 33.5% (3.8%) of the patients who received ibrutinib and in 14.8% (1.9%) of those who received placebo (supplemental Table 5).
When a 78-year old male patient with concomitant use of rivaroxaban reported a subdural hematoma during the second cycle of ibrutinib, the protocol was amended to prohibit the concomitant use of direct Xa-inhibitors, and physicians were educated on CYP3A4 drug-drug interactions. Following this amendment, the incidence of bleeding events per month dropped in the ibrutinib group from 7.9% (95% CI, 4.8%-13.2%) to 1.7% (95% CI, 1.1%-2.6%) and remained nearly stable from 0.93% (95% CI, 0.4%-2.4%) to 0.85% (95% CI, 0.5%-1.5%) in the placebo group (supplemental Table 7).
Cardiac arrhythmias occurred at any grade in 34 (21.5%, 21 [13.3%] grade 1-2) patients in the ibrutinib group and in 12 (7.7%, 10 [6.5%] grade 1-2) patients in the placebo group. One case of ventricular arrhythmia was reported in the ibrutinib group. The event occurred in a 70-year-old woman who developed QT-prolongation and ventricular arrhythmia after 2 completed cycles of ibrutinib, resulting in an impaired left ventricular ejection function. The patient died 3 years later from severe dilatative cardiomyopathy.
Hypertensive disorders occurred at any grade in 18 (11.4%) patients receiving ibrutinib and 7 (4.5%) patients receiving placebo. The incidence of grade 3 or higher hypertensive disorders was the same in both groups (3 patients [1.9%] each).
Four patients in the ibrutinib group and 5 patients in the placebo group had a fatal AE. The causes of death were Escherichia coli sepsis, pneumonia, cardiovascular failure, diffuse large B-cell lymphoma, ovarian cancer, and unknown (n = 4). Second cancers were reported in 17 of 158 patients (10.8%) treated with ibrutinib and in 24 of 155 patients (15.5%) treated with placebo (supplemental Table 8). Richter’s transformation occurred in no patient in the ibrutinib group and 1 patient in the placebo group.
Discussion
CLL12 is the first and largest trial evaluating targeted drugs in treatment-naïve, early-stage patients with CLL. The randomized, double-blind, placebo-controlled phase 3 trial compared treatment with ibrutinib to placebo in asymptomatic patients with Binet stage A CLL at increased risk of progression defined by the GCLLSG score. After a median follow-up of 31 months, the primary endpoint analysis confirmed the efficacy of ibrutinib in prolonging time to active disease progression and initiation of subsequent CLL treatment in patients with early-stage CLL. Due to an insufficient number of events, an analysis of survival cannot be performed at the time of the publication, and the CLL12 trial continues. As the CLL12 trial studied a rather indolent and young subgroup of CLL patients, this results in a long observation period to assess the endpoint of survival.
The risk of progression of early-stage CLL was determined by the GCLLSG score, which categorizes 4 risk groups with different survival using 8 clinical and laboratory markers.13 The score incorporates some parameters that are not widely available, like serum thymidine kinase. Therefore, a simplified prognostic score was created, the CLL-IPI, which has become an internationally accepted stratification tool for patients with CLL.11 When applying the CLL-IPI to the CLL12 treatment cohort, only 46% of the randomized patients who were at intermediate to high risk with the GCLLSG score showed an intermediate to very high risk as defined by the CLL-IPI (supplemental Table 3; supplemental Figure 3).
Study assumptions of the CLL12 trial were based on the external data of the GCLLSG CLL1, CLL4, and CLL8 study containing patients with Binet stage A that were initially observed (supplemental Figure 1). In the CLL12 trial, the median EFS of the placebo group was remarkably longer. A possible explanation for the unexpected deviation of the initial study assumption may have been the protocol specification to continue treatment until symptomatic progression with treatment indication as defined per iwCLL guidelines and ensured by central medical review of response data. Another reason may have been the double-blind, placebo-controlled study design, where physicians may have been more hesitant and patients less insisting on administering the next CLL treatment.
It should be pointed out that interpretation of survival will be confounded by the failure to control and mandate ibrutinib as the first anti-CLL therapy in the placebo group. The ideal comparison would have been immediate vs delayed ibrutinib therapy. At the time of CLL12 initiation, ibrutinib was not approved; therefore, we hypothesized that early use of ibrutinib would delay time to chemoimmunotherapy, thereby delaying chemotherapy-associated side effects and ultimately translating into a long-term survival benefit. The nonavailability of ibrutinib might also have contributed to the high recruitment of younger patients, which may have impacted the results. The approval for ibrutinib as first-line treatment in CLL followed 3 years later and was based on data from the randomized, multicenter, open-label phase 3 RESONATE-2 study demonstrating significant improvement on all efficacy endpoints when compared with chlorambucil in treatment-naïve patients.3 With novel drugs or combinations being approved, like venetoclax combined with obinutuzumab, some patients will receive these therapies as first-line therapy.6 Given this dynamic progress in the field, it was impossible to control the first-line therapy for time periods extending over several years. This illustrates a key challenge of clinical trials in early-stage CLL, as the low event rate often results in a long observation period that may affect the value of a highly innovative study design at the time of initiation.
So far, early intervention studies have failed to demonstrate a survival benefit when comparing early chemotherapy-based interventions to observation as an established standard of care. In therapeutic studies performed by the French Cooperative Group, immediate chlorambucil compared with deferred treatment did not translate into a survival benefit in Binet stage A CLL patients.18 In the untreated group of the first trial, almost 50% of patients did not have progression to more advanced disease and did not need therapy after a follow-up of more than 11 years; however, 27% of patients died of causes related to the disease. The CLL1 and CLL7 trials of the German CLL Study Group and French Cooperative Group on CLL used a risk-stratified design including established risk factors of that time to identify early-stage patients at increased risk for progression and compared chemo- (fludarabine-mono in the CLL1 trial) and chemoimmunotherapy (fludarabine, cyclophosphamide, and rituximab in the CLL7 trial) to observation.19,20 With a follow-up of more than 8 years (CLL1 trial) and more than 4 years (CLL7 trial), therapeutic interventions delayed time to disease progression but so far did not translate into a survival benefit.
The rationale for postponing treatment has been to prevent drug-related side effects and to avoid the oncogenic potential of chemotherapy-based interventions.18,21 The use of CLL-directed treatment in early-stage CLL should exclusively be limited to controlled clinical trials. In the CLL12 trial, the majority of placebo patients have not been treated. A critical assessment will be performed considering the effects of a potential overtreatment, including delayed side effects as well as a potential selection of resistant clones that may alter the response to subsequent CLL treatment.22 Therefore, the CLL12 trial will include a molecular follow-up including testing for BTK or PLC γ mutations for all ibrutinib refractory patients.
A highly relevant finding of the primary endpoint analysis of the CLL12 protocol is the first comparison of ibrutinib vs placebo therapy, providing the cleanest data set for undesired events and for ibrutinib toxicity so far. Surprisingly, the overall incidence and severity of AEs were similar in the 2 treatment groups. Ibrutinib caused a significantly higher incidence of cardiovascular and bleeding events compared with placebo. Bleeding events occurred in more than 30% of ibrutinib-treated patients, although rarely of higher CTC-grade. In the pivotal phase 3 trials that led to the approval of ibrutinib, severe bleeding complications were reported in up to 2% of ibrutinib-treated patients.2,4 An integrated, retrospective analysis of 15 ibrutinib trials revealed a low-grade bleeding rate of 35% (as compared with 15% in the comparator arms), whereas severe hemorrhage occurred in 4.4% of ibrutinib-treated patients (as compared with 2.8% in the comparator arms).23 Use of anticoagulants and/or antiplatelets in this study was common and increased the risk for major hemorrhage in ibrutinib-treated patients. Contrary to the general recommendations, we amended the protocol to prohibit the simultaneous use of direct Xa inhibitors in the CLL12 protocol and trained the treating physicians about CYP3A4 interactions. These measures resulted in a remarkable decline of bleeding episodes in ibrutinib-treated patients.
Cardiac arrhythmias occurred in more than 20% of patients in the ibrutinib group, mostly atrial fibrillation of lower CTC grade. In line with previous reports of ventricular dysfunction in trials studying ibrutinib, 1 case of ventricular tachycardia was reported in the ibrutinib group of the CLL12 protocol.24,25 Second-generation BTK inhibitors demonstrate a greater BTK selectivity and less off-target inhibition as compared with ibrutinib.26,27 The first direct comparison of the less selective inhibitor acalabrutinib vs ibrutinib demonstrated noninferior PFS with fewer cardiovascular events, including a significantly lower rate of atrial fibrillation.28 A pooled analysis reported cardiac AEs in 17% of all acalabrutinib-treated patients, and the most common cardiac AE was atrial fibrillation in 5%.29 Further evaluation of BTK inhibitor-associated cardiotoxicity, as well as interactions with cardiotoxic drugs, will be a key requirement of future studies.
The distribution of AEs in the placebo group appeared to represent the typical health- and leukemia-related problems, most commonly pneumonia, basal cell carcinoma, and acute myocardial infarction. The results indicate that secondary complications may be triggered by reduced immune surveillance in untreated CLL and point to a potential protective effect of ibrutinib preventing thromboembolic complications by decreasing glycoprotein VI collagen-mediated platelet activation, spreading, and aggregation.30
In our opinion, while the CLL12 trial met its primary endpoint, the lack of a demonstrated survival benefit, due to pending survival analysis, does not justify to suggest changing the current recommendation of a “watch and wait” strategy in asymptomatic, early-stage patients until progression to advanced stages or occurrence of CLL-associated symptoms. Modifications of this observational approach require evidence of a significant survival advantage in the ibrutinib arm.
The CLL12 results confirm the cardiovascular toxicity of ibrutinib. The high incidence of AEs observed in the placebo group demonstrated that even early stage, untreated CLL may be associated with relevant morbidity. Survival analysis will clarify whether an early therapeutic intervention reduces the risk of secondary complications which contribute to CLL-related morbidity and mortality.
Acknowledgments
The authors thank all patients, family members, and study staff. A special thanks goes to the CLL12 team of the German CLL Study Group Office including Jasmin Bahlo, Christina Rhein, Henrik Gerwin, Tanja Annolleck, Marie Kronmüller, Jan-Erik Mittler, and Olga Korf. A special thanks goes to the Data Safety and Monitoring Board members Ulrich Jäger, Daniel Catovsky, Tadeusz Robak, and Martin Hellmich, who have ensured patient safety through their continuous efforts.
This trial was supported by Janssen-Cilag.
The Data Safety and Monitoring Board is an independent group of scientists including Ulrich Jäger (Austria), Daniel Catovsky (United Kingdom), Tadeusz Robak (Poland), and Martin Hellmich (Germany).
The funders of the study approved the study design and had the opportunity to review the manuscript but had no role in data collection, data analysis, or data interpretation and had no influence on the content of the manuscript.
Authorship
Contribution: P.L. and M.H. provided primary authorship, trial design, input, and data interpretation; C.Z. and S.R. performed statistical analysis for the trial design, analyzed and interpreted the data; and P.L., P.C., M.F., O.A.-S., J.v.T., A.-M.F., K.-A.K., U.V.-K., E.T., L.M., M.J.E., R.S., W.F., T.G., C.B., M.R., M.S., C.-M.W., K.F., S.S., B.E., and M.H. enrolled patients, performed the research, and contributed to the manuscript.
Conflict-of-interest disclosure: P.L. reports grants and other from Janssen-Cilag, and other from Abbvie and F. Hoffman-LaRoche during the conduct of the study. P.C. reports other from AbbVie, F. Hoffman-LaRoche, Gilead, Janssen-Cilag, and Mundipharma during the conduct of the study. O.A.-S. reports grants, personal fees, and nonfinancial support from Janssen, AbbVie, and Roche, personal fees and nonfinancial support from Gilead, grants from BeiGene, and personal fees from AstraZeneca outside the submitted work. J.v.T. reports personal fees, nonfinancial support and other from Janssen-Cilag, Roche, and AbbVie, and nonfinancial support from Celgene outside the submitted work. A.-M.F. reports personal fees from Janssen outside the submitted work. E.T. reports grants and personal fees from Roche during the conduct of the study, grants, personal fees and nonfinancial support from Abbvie, personal fees and nonfinancial support from Janssen outside the submitted work. C.B. reports grants and personal fees from Boehringer Ingelheim, Roche, Celgene, and Novartis outside the submitted work. C.-M.W. reports grants and personal fees from Janssen-Cilag during the conduct of the study; grants and personal fees from Janssen-Cilag, Hoffmann-La Roche, AbbVie, and Gilead outside the submitted work. K.F. reports other from Roche and AbbVie during the conduct of the study. S.S. reports grants, personal fees, and nonfinancial support from AbbVie, AstraZeneca, Celgene, Gilead, F. Hoffmann La-Roche, Janssen, and Novartis during the conduct of the study; grants, personal fees and nonfinancial support from AbbVie, AstraZeneca, Celgene, F. Hoffmann La-Roche, Janssen, and Novartis outside the submitted work. B.E. reports grants and personal fees from Janssen-Cilag, Roche, AbbVie, and Gilead, and personal fees from Novartis Celgene, ArQule, AstraZeneca, Oxford Biomedica (UK), and grants from BeiGene outside the submitted work. M.H. reports other from AbbVie, F. Hoffman-LaRoche, Gilead, Janssen-Cilag, and Mundipharma during the conduct of the study. The remaining authors declare no competing financial interests.
Correspondence: Petra Langerbeins, Department I of Internal Medicine & Center of Integrated Oncology Cologne Bonn, University Hospital of Cologne, Kerpener Str 62, 50937 Cologne, Germany; e-mail: petra.langerbeins@uk-koeln.de.
Presented in abstract form at the 15th International Conference on Malignant Lymphoma, Lugano, Switzerland, 18-22 June 2019; at the 24th European Hematology Association Congress, Amsterdam, the Netherlands, 13-16 June 2019; and at the International Workshop on Chronic Lymphocytic Leukemia, Edinburgh, Scotland, 20-23 September 2019.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal