


Abstract
Cholesterol is a vital lipid for cellular functions. It is necessary for membrane biogenesis, cell proliferation, and differentiation. In addition to maintaining cell integrity and permeability, increasing evidence indicates a strict link between cholesterol homeostasis, inflammation, and hematological tumors. This makes cholesterol homeostasis an optimal therapeutic target for hematopoietic malignancies. Manipulating cholesterol homeostasis by either interfering with its synthesis or activating the reverse cholesterol transport via the engagement of liver X receptors affects the integrity of tumor cells both in vitro and in vivo. Cholesterol homeostasis has also been manipulated to restore antitumor immune responses in preclinical models. These observations have prompted clinical trials involving acute myeloid leukemia to test the combination of chemotherapy with drugs interfering with cholesterol synthesis (ie, statins). We review the role of cholesterol homeostasis in hematopoietic malignancies as well as in cells of the tumor microenvironment and discuss the potential use of lipid modulators for therapeutic purposes.
Regulation of cholesterol homeostasis by SREBP2 and LXRs
Cholesterol homeostasis is mainly regulated by 2 families of transcription factors: the sterol regulatory element binding proteins (SREBPs)1 and the liver X receptors (LXRα and β isoforms).2,3 The detailed and comprehensive description of pathways regulating cholesterol homeostasis has been reviewed elsewhere.4-8
SREBP2 activates the transcription of enzymes of the mevalonate pathway, such as 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), the rate-limiting enzyme along the pathway leading to the synthesis of cholesterol and isoprenoids (Figures 1A and 2). HMGCR is the main target of the widely used blood cholesterol–lowering drugs statins.6 In addition, SREBP2 regulates the uptake of cholesterol through the induction of low-density lipoprotein receptor (LDLR) expression.9 On the other hand, LXRs control the reverse cholesterol transport pathway (Figure 1B), through which the excess of cholesterol is returned to the liver for excretion as bile acids.10 Two extreme conditions help to understand how these 2 transcription factors cooperate to maintain the homeostatic control of cellular cholesterol levels. In conditions of low cholesterol levels, SREBP cleavage-activating protein (SCAP),11 an endoplasmic reticulum (ER) protein strictly associated with SREBP2, escorts SREBP2 to the Golgi apparatus. There, it is cleaved by 2 serine proteases (S1P and S2P) generating N-terminal fragments,5 which bind DNA regulatory sites in the promoter of genes encoding cholesterol synthesis enzymes, therefore activating the synthesis of cholesterol (Figure 1A).9 In this condition, LXRs remain in a repressive state. In conditions of high cholesterol levels, the trimolecular complex made up of insulin-induced gene (INSIG), SCAP, and SREBP2 remain in the ER, blocking the activation of SREBP2 and, therefore, cholesterol synthesis (Figure 1B).12 The stable formation of the trimolecular complex in the ER is allowed by binding of oxysterols, such as 25-hydroxycholesterol (25-HC), to INSIG13 and/or by binding of cholesterol or desmosterol to SCAP (Figure 1B).14 This binding induces a conformational change avoiding SCAP-SREBP2 escort to the Golgi apparatus.14 Moreover, in conditions of high cholesterol levels, oxysterols or desmosterol3,14 bind and activate the LXR/RXR heterodimers (Figure 1B), which in turn activate specific LXR target genes, such as ATP-binding cassette transporters A1 and G1 (ABCA1 and ABCG1),15 ultimately promoting the elimination of the excess of cellular cholesterol. 25-HC as well as other oxysterols regulate cholesterol homeostasis both in an SREBP2-dependent and an LXR-dependent manner. Indeed, oxysterols block cholesterol synthesis by inhibiting SCAP-SREBP2 escort to the Golgi apparatus and reduce intracellular cholesterol levels through the LXR-dependent expression of ABCA1 with subsequent cholesterol efflux.13,15 Oxysterols can be generated through enzymatic reactions by means of cholesterol hydroxylases or autooxidation (Figure 2).16,17 Mice knockout for 3 main enzymes involved in the synthesis of oxysterols (ie, Ch25h [25-HC], Cyp46a1 [24S-HC], and Cyp27a1 [27-HC]), have altered LXR responses to dietary cholesterol (Figure 2).18 In accordance with these results, in vivo enforced expression of sulfotransferase 2B1b (SULT2B1b), an enzyme inactivating oxysterols by sulfation,19 recapitulates the metabolic phenotype of triple knockout mice,18 thus adding a further layer of complexity to LXR and cholesterol regulation. Although the role played by SULT2B1b in interference with LXR activity has recently emerged, how sulfate oxysterols interfere with LXR signaling is still puzzling. Sulfate oxysterols are hydrophilic and therefore can be more easily depleted from the cells,20 thus reducing intracellular oxysterols and upregulating cholesterol uptake through LDLR.20 This pathway is also expected to increase the SREBP2-dependent cholesterol biosynthesis.21 Sulfate oxysterols have been also shown to antagonize LXR activity.22 These data are corroborated by experiments showing that the sulfated form of 24S-HC oxysterol displays an LXR affinity four fold higher than the nonsulfated form.22 Altogether, these results point to a dual role exerted by SULT2B1b in LXR inhibition (ie, through SREBP2 activation and LXR antagonism).
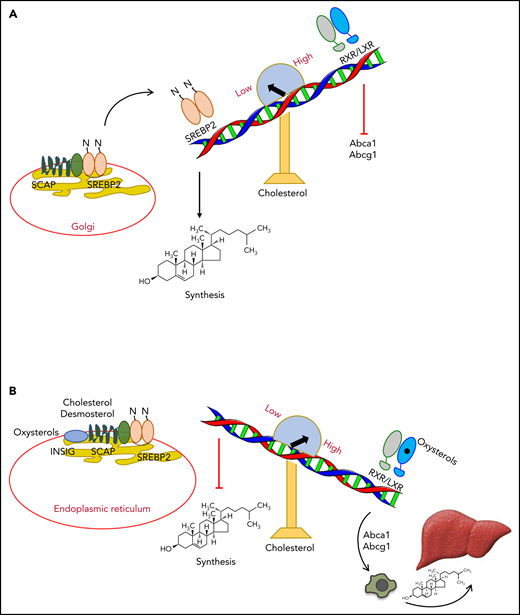
Regulation of cholesterol homeostasis by SREBP2 and LXR. (A) In conditions of low cholesterol levels, SCAP escorts SREBP2 to Golgi apparatus, where it is cleaved and activated. Subsequently, SREBP2 binds and activates the expression of genes regulating de novo cholesterol synthesis. LXRs remain in a repressive state. (B) In conditions of high cholesterol levels, oxysterols, cholesterol, or desmosterol keeps the trimolecular complex INSIG/SCAP/SREBP2 in the ER, not allowing cholesterol synthesis. Oxysterols engage LXRs, which activate expression of the cholesterol transporters ABCA1 and ABCG1. These transporters induce the reverse cholesterol transport pathway, leading to elimination of excess cholesterol through bile acid formation and excretion.
Regulation of cholesterol homeostasis by SREBP2 and LXR. (A) In conditions of low cholesterol levels, SCAP escorts SREBP2 to Golgi apparatus, where it is cleaved and activated. Subsequently, SREBP2 binds and activates the expression of genes regulating de novo cholesterol synthesis. LXRs remain in a repressive state. (B) In conditions of high cholesterol levels, oxysterols, cholesterol, or desmosterol keeps the trimolecular complex INSIG/SCAP/SREBP2 in the ER, not allowing cholesterol synthesis. Oxysterols engage LXRs, which activate expression of the cholesterol transporters ABCA1 and ABCG1. These transporters induce the reverse cholesterol transport pathway, leading to elimination of excess cholesterol through bile acid formation and excretion.
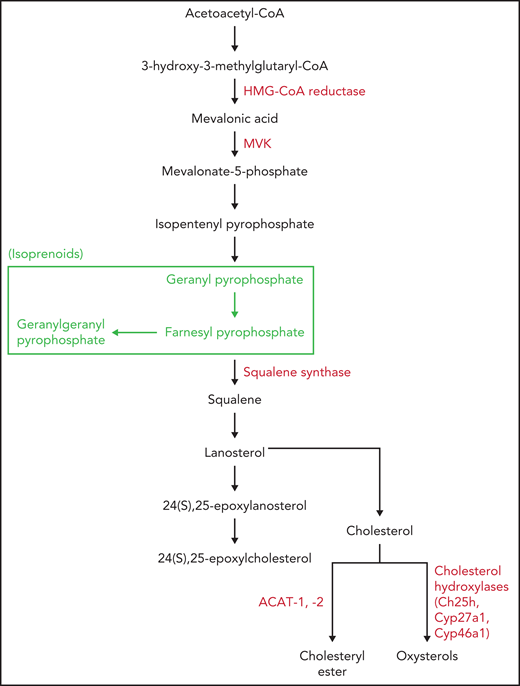
Cholesterol, CE, and oxysterol synthesis. The main enzymes involved in cholesterol, CE, and oxysterol synthesis (red). HMG-CoA reductase, squalene synthase, and ACAT-1 enzymes are also the target of specific drugs. The pathway leading to isoprenoid formation is shown (green).
Cholesterol, CE, and oxysterol synthesis. The main enzymes involved in cholesterol, CE, and oxysterol synthesis (red). HMG-CoA reductase, squalene synthase, and ACAT-1 enzymes are also the target of specific drugs. The pathway leading to isoprenoid formation is shown (green).
Manipulating cholesterol homeostasis in hematopoietic malignancies
The role played by cholesterol in cell membrane integrity and cell proliferation9 predicts the dampening of leukemia survival and expansion through the manipulation of cholesterol homeostasis. Whether modulating SREBP2 or LXR pathways promotes similar or different effects is the object of active investigation in the hematology field, with the aim to identify new therapeutic targets.
Early observations showed that in contrast to normal cells, leukemic cells require higher cholesterol levels either by increasing synthesis or by increasing the uptake.23 Increased cholesterol requirements would fulfill the high rate of proliferation of leukemic cells, improve survival, or protect cells from therapy. As a consequence and contrary to normal myeloid cells in the bone marrow, primary acute myeloid leukemia (AML) samples and AML cell lines underwent apoptosis after mevastatin treatment, indicating involvement of the mevalonate pathway in the maintenance of leukemic cell survival (Figure 3A; Table 1).24 Similar results were also reported for lovastatin, which was toxic to primary AML cell samples and cell lines but not to acute lymphocytic leukemia samples.25 In accordance with protective effects of cholesterol to therapies, the exposure of primary AML samples to the chemotherapeutic agents cytarabine or daunorubicin induced much greater cellular cholesterol levels than those seen in similarly treated normal bone marrow samples.26 In 75% of AML samples treated with daunorubicin or cytarabine, cholesterol increments were associated with higher levels of mRNAs encoding HMGCR and the cholesterol-importing LDLR.27 The treatment of AML cell lines with mevastatin or zaragozic acid A (ZAA) (Figure 3A; Table 1)28 was able to contrast the previously noted cholesterol increments and induce apoptosis of cells undergoing radio- and chemotherapy.26 The induction of leukemic cell apoptosis involving the targeting of the rate-limiting enzyme HMGCR was also observed in multiple myeloma (MM) cells treated with atorvastatin or fluvastatin (Table 1).29 In this study, the addition of dipyridamole, a compound that blocks the feedback response (ie, the upregulation of HMGCR and HMG-CoA synthase 1), in vitro and in vivo in leukemia xenografts following statin treatment further synergized with statins to enhance apoptosis of MM, AML cell lines, and primary patient cells. Of note, dipyridamole was shown to inhibit the cleavage of SREBP2, the main component of the feedback response.29
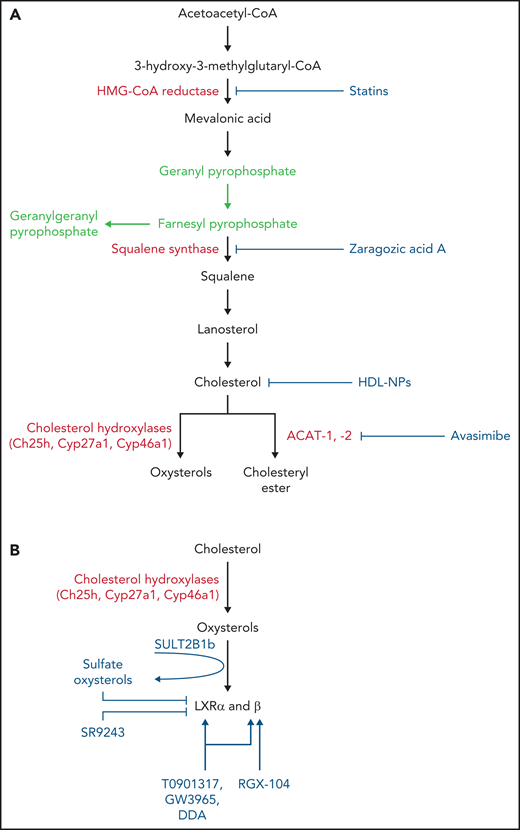
Drugs interfering with cholesterol and CE synthesis and LXR activity. (A) Drugs interfering with cholesterol and CE synthesis. Statins block both cholesterol synthesis and isoprenoid formation (green), whereas ZAA blocks cholesterol synthesis, leaving intact isoprenoid formation (green). Avasimibe blocks formation of CEs by inhibiting the ACAT-1. HDL-NPs promote cellular cholesterol efflux and restrain cholesterol delivery by targeting scavenger receptor type B-1. (B) LXRs can be inhibited by sulfate oxysterols through the activity of sulfotransferase 2B1b (SULT2B1b) enzyme and by the inverse agonist SR9243. LXRs are activated by oxysterols generated through the activity of cholesterol hydroxylases (eg, ch25h, Cyp27a1, Cyp46a1) or auto-oxidation and by LXRβ-selective (RGX-104) and nonselective LXR agonists (T0901317, GW3965, DDA).
Drugs interfering with cholesterol and CE synthesis and LXR activity. (A) Drugs interfering with cholesterol and CE synthesis. Statins block both cholesterol synthesis and isoprenoid formation (green), whereas ZAA blocks cholesterol synthesis, leaving intact isoprenoid formation (green). Avasimibe blocks formation of CEs by inhibiting the ACAT-1. HDL-NPs promote cellular cholesterol efflux and restrain cholesterol delivery by targeting scavenger receptor type B-1. (B) LXRs can be inhibited by sulfate oxysterols through the activity of sulfotransferase 2B1b (SULT2B1b) enzyme and by the inverse agonist SR9243. LXRs are activated by oxysterols generated through the activity of cholesterol hydroxylases (eg, ch25h, Cyp27a1, Cyp46a1) or auto-oxidation and by LXRβ-selective (RGX-104) and nonselective LXR agonists (T0901317, GW3965, DDA).
Effects of drugs interfering with cholesterol homeostasis on tumor and tumor-infiltrating immune cells
| Drug | Target | Activity on tumor cells | Activity on immune cells |
|---|---|---|---|
| Statins (mevastatin, lovastatin, atorvastatin, fluvastatin, rosuvastatin, simvastatin) | HMGCR, Isoprenoid formation | Apoptosis of leukemic cells (AML) and myeloma cells (MM) | Blocking Rab5 prenylation leading to prolonged Ag presentation |
| ZAA | Squalene synthase | Apoptosis of leukemic cells (AML) | Increase CD11c+ DC activity, decrease of protumor neutrophils |
| T0901317 | LXR agonist | Apoptosis of leukemic cells (CLL and BPDCN) | |
| GW3965 | LXR agonist | Apoptosis of BPDCN cells | Expression of NK cell-activating ligands MICA and MICB and suppression of MDSCs |
| DDA | LXR partial agonist | Lethal autophagy of leukemic cells (AML) | Increased infiltration of CD3+ T cells and CD11c+ DCs |
| Avasimibe | ACAT-1 inhibitor | Suppression of leukemic cell proliferation (CML) | Enhanced TCR clustering and immunological synapse of CD8+ T cells |
| HDL-NP | Scavenger receptor type B-1 | Apoptosis of DLBCL cells | |
| SR9243 | LXR inverse agonist | Apoptosis of tumor cells | Increased CD11c+ DC and CD8+ T-cell activity |
| RGX-104 | LXRβ agonist | Suppression of MDSCs | |
| SULT2B1b | LXR antagonist | Increased CD11c+ DC activity, decrease in protumor neutrophils |
| Drug | Target | Activity on tumor cells | Activity on immune cells |
|---|---|---|---|
| Statins (mevastatin, lovastatin, atorvastatin, fluvastatin, rosuvastatin, simvastatin) | HMGCR, Isoprenoid formation | Apoptosis of leukemic cells (AML) and myeloma cells (MM) | Blocking Rab5 prenylation leading to prolonged Ag presentation |
| ZAA | Squalene synthase | Apoptosis of leukemic cells (AML) | Increase CD11c+ DC activity, decrease of protumor neutrophils |
| T0901317 | LXR agonist | Apoptosis of leukemic cells (CLL and BPDCN) | |
| GW3965 | LXR agonist | Apoptosis of BPDCN cells | Expression of NK cell-activating ligands MICA and MICB and suppression of MDSCs |
| DDA | LXR partial agonist | Lethal autophagy of leukemic cells (AML) | Increased infiltration of CD3+ T cells and CD11c+ DCs |
| Avasimibe | ACAT-1 inhibitor | Suppression of leukemic cell proliferation (CML) | Enhanced TCR clustering and immunological synapse of CD8+ T cells |
| HDL-NP | Scavenger receptor type B-1 | Apoptosis of DLBCL cells | |
| SR9243 | LXR inverse agonist | Apoptosis of tumor cells | Increased CD11c+ DC and CD8+ T-cell activity |
| RGX-104 | LXRβ agonist | Suppression of MDSCs | |
| SULT2B1b | LXR antagonist | Increased CD11c+ DC activity, decrease in protumor neutrophils |
Ag, antigen; MDSC, myeloid-derived suppressor cell.
The blockade of HMGCR by statins inhibits not only the synthesis of cholesterol but also the generation of the isoprenoids geranylgeranyl pyrophosphate and farnesyl pyrophosphate (Figure 2).30 Because prenylation of GTPases, such as Ras, is critically involved in protein activation, previous studies have attempted to evaluate whether the antileukemic effects of statins could be due to the blockade of Ras isoprenylation.31 Lovastatin-induced apoptosis of human AML cells was shown to be dependent on the blockade of Ras isoprenylation.32 However, simultaneous inhibition of geranylgeranyl transferase and farnesyltransferase activities proved to be not very active, indicating that the antileukemic effect of statins could require a mechanism independent of or complementary to Ras prenylation,31 as further indicated by the observation of statin-induced apoptosis in primary AMLs harboring Ras activated by “hotspot” mutations.24 In addition, the induction of apoptosis of leukemic cells treated with ZAA would argue against an antileukemic effect of statins mediated by Ras isoprenylation because ZAA blocks cholesterol synthesis, leaving intact the generation of isoprenoids (Figure 3A).26,30 By applying systems biology approaches to coculture models recapitulating chronic lymphocytic leukemia (CLL) microenvironment, research showed that simvastatin decreased CLL cell survival and proliferation as well as cell adhesion.33 Simvastatin also enhanced the proapoptotic, antitumor activity of the BCR signaling inhibitor ibrutinib and the B-cell lymphoma-2 inhibitor venetoclax,33 the latter combination already being shown to affect various primary leukemia and lymphoma cells but not normal peripheral blood mononuclear cells.34 Of note, the addition of geranylgeranyl pyrophosphate rescued cell viability, indicating a possible contribution of protein prenylation to leukemia survival.34 From a clinical standpoint, high-dose pravastatin in combination with idarubicin and intermediate dose cytarabine was explored in phase 1 and 2 trials in patients with relapsed AML. Although the clinical response was high, the study included patients with AML who had a favorable risk.35 The subsequent phase 2 clinical trial in patients with poor-risk AML gave encouraging but not definitive results.36
Cholesterol biosynthesis is transcriptionally induced in AML cells under hypoxic stress conditions and attenuated by cytarabine and quizartinib, the latter being a selective inhibitor of class III receptor tyrosine kinases, including FMS-related tyrosine kinase 3.37 Interestingly, cytarabine increases intracellular cholesterol content in AML cells independently of O2 tension, possibly switching to scavenger mode through the upregulation of CD36. In this setting, rosuvastatin exerts a significant antileukemic activity against a wide variety of AML cells and acts synergistically with cytarabine at different levels of O2, an observation deserving investigation of rosuvastatin-based therapies to target residual AML cells that reside in low-O2 environments, such as the leukemic niche. Indeed, Golub and colleagues have recently shown in a leukemia-stroma coculture system that lovastatin can affect leukemia stem cells (LSCs) while sparing normal hematopoietic stem and progenitor cells.38 Lovastatin showed anti-LSC activity in vitro and in vivo via inhibition of HMGCR.38 We hypothesize that the sensitivity of LSCs to lovastatin reflects the upregulation of cholesterol synthesis under hypoxic conditions of the leukemic niche. Lovastatin was also shown to dampen in a heterogenous manner the primitive subfraction of CD34+ AML cells.39 CD34+ AML cells that poorly responded in vitro identified a subgroup of AML patients with poor prognosis.39
Apoptotic cell death is also a distinctive feature of LXR activation in CLL B and T cells.40 Whereas in T-CLL cells, now called T-cell prolymphocytic leukemia, apoptosis is preceded by the blockade of interleukin (IL) 2-dependent proliferation through inhibition of retinoblastoma protein phosphorylation and decreased expression of the cyclin B, in B-CLL cells it is associated with the inhibition of bcl-2 and MMP-9 expression.40 Apoptotic cell death is also induced in primary blastic plasmacytoid dendritic cell neoplasm (BPDCN) cells and BPDCN cell lines undergoing LXR agonist treatment both in vitro and in vivo.41 In this context, LXR-induced apoptotic cell death occurs through inhibition of NF-κB activation as well as Akt and STAT5 phosphorylation in response to IL-3. Of note, transcriptomics of BPDCN primary cells confirmed the dysregulation of genes involved in cholesterol homeostasis, particularly LXR target genes.41
Leukemic cells undergoing statin-induced cholesterol inhibition can activate autophagy, which exerts a cytoprotective role. Indeed, the suppression of autophagy by pharmacologic or genetic treatments induces apoptosis of statin-treated leukemic cells.42 The treatment of primary AML samples with dendrogenin A (DDA), a newly discovered cholesterol metabolite that is a partial LXR agonist, induces lethal autophagy in vitro and in vivo. DDA also inhibits the cholesterol biosynthesizing enzyme 3β-hydroxysterol-Δ8,7-isomerase (D8D7I), leading to sterol accumulation and cooperating in autophagy induction.43 These effects are not observed with canonical LXR agonists such as the oxysterol 22(R)-hydroxycholesterol or the synthetic agonists T0901317 and GW3965 (Figure 3B;,Table 1).44 The combination of DDA and idarubicin synergistically enhanced the autophagic cell death in AML cells, maximizing DNA damage induced by idarubicin and decreasing DNA repair.45 Differently from the results reported in CD34+ AML cells treated with lovastatin,39 the induction of lethal autophagy by the LXR agonist DDA occurred independently of their cytogenetic subgroups and did not distinguish bulk cancer cells from cancer cell progenitors.44
Intracellular cholesterol may undergo esterification, a process mediated by acetyl-coenzyme A cholesterol acetyltransferase 1 and 2 (ACAT-1 and -2), which allows for cholesterol storage (Figure 2).9 CML cells undergo an aberrant accumulation of cholesteryl ester (CE), possibly due to the BCR-ABL translocation. Interestingly, the blockade of cholesterol esterification with avasimibe, a potent inhibitor of ACAT-1 (Figure 3A; Table 1), suppressed CML cell proliferation in BCR-ABLT315I mutation and in cells resistant to imatinib.46 This effect was due to the avasimibe-induced downregulation of the MAPK pathway, rendering CML cells sensitive to imatinib treatment in vitro and in vivo.46
High-density lipoproteins (HDLs) target the scavenger receptor type B-1. Synthetic HDL nanoparticles (HDL-NPs), which mimic natural HDLs, have been successfully used to alter cholesterol homeostasis and induce apoptosis of lymphoma cells, particularly diffuse large B-cell lymphoma (DLBCL) cells showing increased cholesterol biosynthesis (Figure 3A; Table 1).47 The treatment of mice bearing B-cell lymphoma xenografts with HDL-NP inhibited B-cell lymphoma growth.47
In BCR-dependent DBLCL cells, SYK blockade decreased membrane cholesterol content and inhibited BCR capping.48 Moreover, the combination of HDL-NP with the SYK inhibitor R406 or ibrutinib induced cellular cholesterol reduction and apoptosis of resistant activated B-cell DLBCL cells.49 This treatment could be applied to patients with non-GC DLBCL receiving rituximab-cyclophosphamide, hydroxydaunorubicin, vincristine sulfate (Oncovin), and prednisone therapy concomitantly to statins, for whom a worse prognosis was reported.50 Of note, statin-mediated cholesterol depletion was shown to induce CD20 conformational changes affecting the binding of anti-CD20 mAb.51
Altogether, these results indicate that the manipulation of cholesterol homeostasis through the inhibition of cholesterol synthesis, activation of the reverse cholesterol transport, inhibition of cholesterol esterification, or targeting of scavenger receptor type B-1 can be exploited for clinical purposes to enhance the efficacy of antileukemic compounds. The results reported so far also suggest combining drugs targeting both cholesterol pathways to maximize antitumor effects and avoid mechanisms of resistance.
SREBP2 and LXRs at the interface of cholesterol homeostasis and inflammation
Recent studies have highlighted the key role exerted by SREBP2 and LXRs to integrating metabolic and inflammatory signaling. The activation of LXRs in macrophages leads to the inhibition of the expression of certain proinflammatory molecules, such as tumor necrosis factor-α, IL-1β, inducible nitric oxide synthase, IL-6, COX-1, MMP9, CCL2, and CCL7 (Figure 4).52,53 On the other hand, SREBP2 activates inflammasome, ultimately promoting IL-1β release.54 Different mechanisms and players, of which some are still debated, operate in the LXR- and SREBP2-induced regulation of inflammation. In this section, we discuss how SREBP2 and LXRs regulate inflammation focusing on pathways exploitable to restore antitumor immune responses. The detailed and comprehensive description of LXR- and SREBP2-induced regulation of inflammation has been reviewed elsewhere.21,52-56

SREBP2 and LXR pathways regulating inflammation. Upregulation of cholesterol synthesis by SREBP2 also involves NLRP3 activation as SCAP forms a ternary complex and escorts both proteins to the Golgi apparatus (1). NLRP3 and inflammasome activation leads to IL-1β release. Increased cholesterol synthesis induces mitochondrial damage, release of mitochondrial DNA, and activation of the AIM2 component of the inflammasome (2). Oxysterols, such as 25-HC, inhibit inflammation by blocking cholesterol synthesis and inflammasome activation and activating LXRs through multiple mechanisms (3). Inflammasome activation is blocked by inhibition of cholesterol synthesis and avoidance of SCAP/SREBP2 and NLRP3 translocation to the Golgi apparatus. Cholesterol synthesis also leads to formation of GGPP and FPP isoprenoids, which induce prenylation and activation of RhoA, Ras, Rac, Cdc42, and Rab5 small GTPases, which are involved in inflammasome activation, antigen presentation, and cell movement (4). FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate.
SREBP2 and LXR pathways regulating inflammation. Upregulation of cholesterol synthesis by SREBP2 also involves NLRP3 activation as SCAP forms a ternary complex and escorts both proteins to the Golgi apparatus (1). NLRP3 and inflammasome activation leads to IL-1β release. Increased cholesterol synthesis induces mitochondrial damage, release of mitochondrial DNA, and activation of the AIM2 component of the inflammasome (2). Oxysterols, such as 25-HC, inhibit inflammation by blocking cholesterol synthesis and inflammasome activation and activating LXRs through multiple mechanisms (3). Inflammasome activation is blocked by inhibition of cholesterol synthesis and avoidance of SCAP/SREBP2 and NLRP3 translocation to the Golgi apparatus. Cholesterol synthesis also leads to formation of GGPP and FPP isoprenoids, which induce prenylation and activation of RhoA, Ras, Rac, Cdc42, and Rab5 small GTPases, which are involved in inflammasome activation, antigen presentation, and cell movement (4). FPP, farnesyl pyrophosphate; GGPP, geranylgeranyl pyrophosphate.
It has been reported that the inflammasome-dependent release of mature IL-1β requires an increase of intracellular levels of cholesterol, which in turn, by damaging mitochondria, favors mitochondrial DNA release in the cytosol and activation of the AIM2 component of the inflammasome (Figure 4).57 As observed in Ch25h−/− macrophages, the increase of intracellular levels of cholesterol is regulated by SREBP2 activity and cholesterol 25-hydroxylase (CH25H)/25-HC oxysterol axis. Moreover, addition of the oxysterol 25-HC to lipopolysaccharide-activated macrophages blocks inflammasome activation through SREBP2 inhibition (Figure 4).55,57 In agreement with the cholesterol-dependent inflammasome activation, dendritic cells (DCs) deficient for the LXR target genes ABCA1 and ABCG1 accumulate intracellular cholesterol, which in turn induces SREBP2-independent, NLRP3-dependent inflammasome activation.58 Whether this mechanism requires the activation of the AIM2 component of the inflammasome or a different pathway remains unknown. Interestingly, DC activation was also observed in Lxrαβ−/− mice fed a high-cholesterol diet.59 In this context, the activation of DCs mediated by cholesterol loading induced T- and B-cell priming, leading to autoantibody formation and autoimmunity.59 This phenotype could be also fostered by a defective phagocytic clearance of apoptotic cells due to the lack of expression of the membrane receptor Mertk.60
Recently, Guo and colleagues showed a physical association between SCAP-SREBP2 and NLRP3.61 The identification of the molecular link between SCAP-SREBP2 and NLRP3 clarifies the apparent paradox of statin-induced inflammation.61 By inhibiting HMGCR, statins block cholesterol synthesis.6 In this condition, one would expect a reduction of the inflammatory state of monocytes/macrophages. Instead, macrophages pretreated with statins release IL-1β soon after 6 hours of incubation with lipopolysaccharide.61 The results by Guo and colleagues indicate that the blockade of cholesterol synthesis mediated by statins promotes SCAP-SREBP2 translocation and NLRP3 inflammasome activation. Under these conditions, SCAP escorts both SREBP2 and NLRP3 through the formation of a ternary complex (Figure 4).61 The blockade of SCAP-SREBP2 ER-to-Golgi apparatus translocation suppressed NLRP3 inflammasome activation and IL-1β production.61 In macrophages, the activation of NLRP3 by SCAP-SREBP2 translocation is also promoted by the cytokine tumor necrosis factor-α, which induces SREBP2 activity through binding to inflammatory genes.62
Results similar to those observed in vitro by treating macrophages with statins and leading to a hyperinflammasome phenotype are observed in individuals affected by pyrin and mevalonate kinase deficiency (MVK) (Figure 1), leading to familial Mediterranean fever and hyperimmunoglobulin D syndrome.63,64 Inflammasome activation in MVK-deficient cells is the result of impaired prenylation of RhoA/K-ras, resulting in activation of pyrin and subsequent recruitment of the adaptor protein Asc and the protease caspase-1.63 Activated caspase-1 cleaves pro–IL-1β into active IL-1β, thus driving inflammatory pathology in MVK-deficient patients.63,64 While Park and colleagues attributed the inflammatory pathology to impaired RhoA prenylation,63 Akula and colleagues showed defective K-ras prenylation and subsequent loss of PI(3K)-dependent inhibition of pyrin.64 Because statins reduce the formation of geranylgeraniol and other intermediates involved in protein prenylation (Figure 2), one can speculate that statins activate inflammasome by impaired prenylation of RhoA/K-Ras as well as promotion of SCAP-SREBP2 translocation induced by cholesterol synthesis inhibition. Whether both effects play a role or 1 of the 2 effects predominates deserves further investigation. Interestingly, the proinflammatory effects played by statins also extend to their capacity to augment antigen presentation of professional antigen-presenting cells. Statins inhibit the geranylgeranylation of small GTPases, including Rab5, in antigen-presenting cells, resulting in arrested endosomal maturation and prolonged antigen retention, ultimately leading to enhanced antigen presentation and T-cell activation (Figure 4).65
Different mechanisms have been identified and characterized to explain the antiinflammatory activity exerted by LXRs,66 such as the transrepression of NF-κB– and AP-1–activated inflammatory genes67,68 and the ABCA1-dependent inhibition of immune receptors signaling (TLR4-MyD88-TRAF6 signaling).69 The ABCA1-dependent inhibition of immune receptors signaling integrates metabolism and inflammation almost exclusively through the remodeling of membrane lipid rafts. Indeed, ABCA1-dependent changes in membrane lipid organization disrupts the recruitment of MyD88 and TRAF6 to rafts blocking MAPK and NF-κB activation after TLR engagement.69 Additional mechanisms involve the nuclear coreceptor-dependent LXR derepression that increases the expression of omega-3 fatty acids and inhibits NF-κB–dependent inflammatory responses.70 These also include SREBP1-dependent production of mono- or polyunsaturated antiinflammatory fatty acids uncoupling NF-κB binding from gene activation71 and a mechanism based on cholesterol efflux and LXR cis-repression involving direct binding of LXR to inflammatory gene enhancers.72 It has been reported that the activation of LXRs induces the expression of the remodeling enzyme lysophosphatidylcholine acyltransferase 3 (Lpcat3).73 Lpcat3 catalyzes the formation of phosphatidylcholine from saturated lysophosphatidylcholines and unsaturated fatty acetyl-CoAs and promotes membrane incorporation of polyunsaturated fatty acids into phospholipids, especially the arachidonoyl-containing phosphatidylcholine.73 This results in the reduction of free arachidonate, the main precursor of lipid mediators of inflammation, such as prostaglandins, leukotrienes, and thromboxanes.74 Moreover, changes in membrane composition regulate inflammatory kinase activation, particularly the c-Src-JNK pathway that further modulates inflammatory reactions.73
The characterization of SULT2B1b as regulator of LXR responses to dietary cholesterol18 has opened the way to investigate how LXR/SULT2B1b axis interferes with immune cells and immune responses. Lxrβ−/− T and B cells undergo hyperproliferation, indicating that LXR behaves as a metabolic checkpoint uncoupling cholesterol homeostasis from cell proliferation.75 In vitro activated LXR-proficient T cells undergo transient expression of SULT2B1b that restrains LXR activation.75 Whether a similar mechanism interfering with the LXR/Lpcat3 axis occurs during the activation of macrophages remains to be established.
Manipulating cholesterol homeostasis in the tumor microenvironment
Behind the direct effects of SREBP2 and LXRs on tumor cells, there is growing evidence that cholesterol homeostasis controls tumors by dampening immune cells.76,77 Ablation of LXR signaling in immune cells, especially myeloid cells, was proven beneficial for tumor-bearing mice. In a mouse model of lymphoma, we demonstrated that activation of LXR signaling inhibits CCR7 expression and DC migration to draining lymphoid organs, dampening antilymphoma immune responses.78 Restoration of antitumor immune responses was observed in Lxrαβ−/− bone marrow-chimera mice as well as in wild-type mice challenged with SULT2B1b-expressing lymphoma cells (Figure 3B; Table 1),78 thus confirming the reciprocal regulation between LXR and SULT2B1b. This hypothesis was further corroborated by immunohistochemistry analyses of lymphoma samples expressing SULT2B1b and showing higher numbers of tumor-infiltrating CD11c+ DCs and CD3+ T cells as compared with mock-expressing lymphoma cells (Table 1).78 These histologic and functional features were also observed in lymphoma-bearing mice treated with the cholesterol-lowering drug ZAA (Table 1).28 ZAA controlled lymphoma growth in immunocompetent mice and potentiated the antitumor effects of adoptive immunotherapy, significantly prolonging the overall survival of lymphoma-bearing mice treated with the combination of ZAA and adoptive cell therapy.79 Along the same lines, recent studies using the LXR inverse agonist SR9243 in mouse models of cancer have reported increased immune responses promoted by DCs and CD8+ T cells and antitumor effects (Figure 3B; Table 1).80,81
In addition to LXR-dependent mechanisms, we have also reported that oxysterols released by lymphomas are able to recruit neutrophils in the tumor microenvironment endowed with protumor functions in an LXR-independent and CXCR2-dependent manner.82 The pharmacologic and genetic inactivation of oxysterols restored antilymphoma immune responses by reducing the number of tumor-infiltrating neutrophils (Table 1).82 In line with the binding of oxysterols to receptors other than LXRs, it should be mentioned that the EBI2/7α,25-dihydroxycholesterol axis drives B cells toward the extrafollicular area.83,84 Mice whose B cells overexpress hEBI2 display a reduction in GC-dependent immune responses and an increased proliferation and upregulation of cellular oncogenes in the CD5+ B1a B-cell subset, a condition resembling late-onset development of human CLL.85
How do active and inactive LXR ligands/oxysterols induce similar antitumor responses? The expression of LXRα and β isoforms by distinct cell types within the tumor microenvironment may explain these results.86 Indeed, tumor-bearing mice treated with the specific LXRβ agonist RGX-104 rejected tumors by reducing the number of myeloid-derived suppressor cells and increasing the number of antigen-specific T cells (Figure 3B; Table 1).87 Indeed, LXRβ seems to be predominantly active in myeloid immature cells,87 other than T cells,75 and stromal and endothelial cells.88 Along the same lines, the use of LXRα- or β-selective agonists89 or the combination of selective agonists and the SR9243 reverse agonist80,81 could enhance therapeutic effectiveness and overcome limitations associated with broader cell- and tissue-specific activities and side effects, especially those related to increased liver lipogenesis and toxicity.76,90 Human MM cell lines and primary malignant plasma cells treated with synthetic LXR agonists express the NK cell-activating ligands major histocompatibility complex class I chain-related molecule A and B (MICA and MICB) consequently to changes of intracellular cholesterol content induced by LXR activation. LXR-treated MM cells display increased sensitivity to NK cell recognition, thus supporting the use of LXR agonists for the treatment of patients affected by MM (Table 1).91 Finally, the partial LXR agonist DDA has been shown to enhance tumor infiltration of CD3+ T lymphocytes and CD11c+ DCs (Figure 3B; Table 1).92 Therefore, investigating the effects of compounds targeting LXR in appropriate preclinical models could shed light on their mechanisms of action and open new avenues for the treatment of hematopoietic malignancies.
Statins activate NLRP3 inflammasome in myeloid cells.61 However, it appears that statins interfere with antitumor immune responses by regulating isoprenoids formation. Indeed, the blockade of HMGCR operated by statins inhibits the formation of the downstream isoprenoids (ie, farnesyl pyrophosphate and geranylgeranyl pyrophosphate). As a consequence, the small GTPases Ras, Rac, Cdc42, and Ras homolog (ρ) do not undergo prenylation, thus unleashing inflammasome activation.30 These results are corroborated by in vitro and in vivo studies with geranylgeranyltransferase type I (GGTase-I) deficient macrophages. These deficient cells accumulated high levels of active GTP-bound Rac1, Cdc42, and RhoA, leading to enhanced production of proinflammatory cytokines.93,94
As a consequence, the inefficient prenylation of the small GTPase Rab5, induced by statins in antigen-presenting cells, results in arrested endosomal maturation, prolonged antigen retention and enhanced antigen presentation, and T-cell generation/activation (Table 1).65 Exploitation of this effect induced by simvastatin in combination with the immune checkpoint blocker anti-PD-1 mAb induces synergistic antitumor effects in preclinical tumor models, suggesting the possibility of exploiting statins for potentiating antitumor immune responses. These results encourage physicians to design trials that combine statins with immunotherapy. In this context, the reduction of the cholesterol synthetic flux induced by statins might further boost the antitumor immune response by activating type I interferon signaling.95
Antitumor immune responses were also obtained in preclinical models by using avasimibe, an inhibitor of the key cholesterol esterification enzyme ACAT1.96 Treatment of tumor-bearing mice with avasimibe led to enhanced effector functions and increased antigen-specific CD8+ T-cell proliferation (Figure 3A).96 Mechanistically, an increase in the plasma membrane cholesterol level of CD8+ T cells was shown, which promoted enhanced T-cell receptor clustering and signaling, as well as more efficient formation of the immunological synapse.96 The combined therapy of avasimibe plus anti-PD-1 mAbs showed better efficacy than monotherapies in controlling tumor progression (Table 1).96 Of note, treatment of human CD19-directed chimeric antigen receptor T cells with avasimibe increased their effector functions. Avasimibe-treated cells killed CD19-expressing leukemic cells more efficiently and secreted higher levels of interferon-γ as compared with untreated chimeric antigen receptor T cells, thus providing a new option for a combination therapy based on the regulation of T-cell cholesterol homeostasis to enhance antitumor effects.97
Prospects for targeting cholesterol homeostasis in the treatment of hematopoietic malignancies
Recent studies have provided evidence that cholesterol is a multifaceted molecule, regulating cell permeability and integrity and inflammatory responses. As such, the manipulation of cholesterol homeostasis has been shown to affect leukemic cell survival and restore antileukemic immune responses.24,26,76,98
The inhibition of cholesterol synthesis by statin treatments combined with chemotherapeutic regimens provided encouraging clinical responses in patients affected by hematopoietic malignancies.35,36 Moreover, the increasing number of preclinical results reporting successful antitumor immune responses when using drugs that interfere with cholesterol homeostasis combined with immune checkpoint blockers65,96 suggests translating this strategy into clinical practice to treat patients affected by hematopoietic malignancies who are undergoing immunotherapy with immune checkpoint blockers or adoptive cell therapy. Regarding the use of statins in combination with immunotherapy, it should be noted that several factors may influence the clinical activity of statins,99 such as their lipophilic or hydrophilic structure. Lipophilic statins are significantly more potent than hydrophilic ones because of their superior cell penetrance.100 However, lipophilic statins have a short half-life (eg, simvastatin, t1/2 = 2-3 hours), indicating that HMGCR activity would rapidly recover, leading to resynthesis of cholesterol and isoprenoids.99,100 Finally, diets containing geranylgeraniol may overcome the blockade of isoprenoid synthesis induced by statins.99 These factors, which are potentially responsible for the failure of some prospective clinical trials,101 should be taken into account when planning statin-based studies.
Because of the complex regulation of cholesterol metabolism and multiple ways that cholesterol interferes with tumor cells and immune responses, targeting cholesterol metabolism for cancer treatment remains challenging. Therefore, dissecting the mechanisms underlying these pathways could unveil new regulatory networks that maintain cholesterol homeostasis and open new frontiers in the treatment of hematopoietic malignancies. While the pharmacologic manipulation of cholesterol homeostasis in preclinical models dysregulates hematopoietic tumor cell growth and activates antitumor immune responses, 26,29,65,79 clinical investigations are needed to evaluate the potential for targeting these pathways in the treatment of patients with cancer.
Acknowledgments
This work was supported by the Italian Association for Cancer Research (AIRC) grants IG 19016 and IG 22737 (V.R.), and IG 19867 (A.B.).
Authorship
Contribution: A.B. and V.R. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vincenzo Russo, San Raffaele Scientific Institute, via Olgettina 58, 20132, Milan, Italy; e-mail: russo.vincenzo@hsr.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal